
Transcriptomic and Proteomic Analysis of Mannitol-metabolism-associated Genes in Saccharina japonica
2020-07-05 07:36:22ShanChiGuoliangWangTaoLiuXuminWangCuiLiuYuemeiJinHongxinYinXinXuJunYu
Shan Chi, Guoliang Wang, Tao Liu*, Xumin Wang*,Cui Liu,Yuemei Jin Hongxin Yin Xin Xu Jun Yu*
1 College of Marine Life Sciences, Ocean University of China, Qingdao 266003, China
2 Qingdao Haida BlueTek Biotechnology Co., Ltd., Qingdao 266003, China
3 CAS Key Laboratory of Genome Sciences and Information, Beijing Key Laboratory of Genome and Precision Medicine Technologies, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing 100101, China
4 University of Chinese Academy of Sciences, Beijing 100049, China
5 Beijing Key Laboratory of Agricultural Genetic Resources and Biotechnology, Beijing Agro-Biotechnology Research Center,Beijing Academy of Agriculture and Forestry Science, Beijing 100097, China
6 College of Life Sciences, Yantai University, Yantai 264005, China
KEYWORDS Mannitol metabolism;Saccharina japonica;RNA-seq;Proteomic analyses;Biochemical characterization
Abstract As a carbon-storage compound and osmoprotectant in brown algae,mannitol is synthesized and then accumulated at high levels in Saccharina japonica(Sja);however,the underlying control mechanisms have not been studied.Our analysis of genomic and transcriptomic data from Sja shows that mannitol metabolism is a cyclic pathway composed of four distinct steps.A mannitol-1-phosphate dehydrogenase (M1PDH2) and two mannitol-1-phosphatases (M1Pase1 and MIPase2)work together or in combination to exhibit full enzymatic properties.Based on comprehensive transcriptomic data from different tissues, generations, and sexes as well as under different stress conditions, coupled with droplet digital PCR (ddPCR) and proteomic confirmation, we suggest that SjaM1Pase1 plays a major role in mannitol biosynthesis and that the basic mannitol anabolism and the carbohydrate pool dynamics are responsible for carbon storage and anti-stress mechanism.Our proteomic data indicate that mannitol metabolism remains constant during diurnal cycle in Sja. In addition, we discover that mannitol-metabolism-associated (MMA) genes show differential expression between the multicellular filamentous (gametophyte) and large parenchymal thallus(sporophyte) generations and respond differentially to environmental stresses, such as hyposaline and hyperthermia conditions.Our results indicate that the ecophysiological significance of such differentially expressed genes may be attributable to the evolution of heteromorphic generations (filamentous and thallus) and environmental adaptation of Laminariales.
Introduction
Mannitol is one of the most common sugar alcohols in nature and is produced by a variety of living organisms, including bacteria,fungi,terrestrial plants,and algae[1,2].The presence of mannitol has been reported in primary endosymbiotic algae(e.g., species belonging to Chlorophyta [3,4] and a few species of Rhodophyta [5,6]), as well as in secondary endosymbiotic Ochrophyta algae (e.g., brown algae [7–9]) and some other stramenopiles (e.g., Olisthodiscus sp. [10]). As one of the primary photosynthetic products and storage compounds in Laminariales [11–13], mannitol can be stored up to 15%–26%of the organism’s dry weight[14,15].Moreover,mannitol fulfills key physiological roles, including protecting against environmental stresses and acting as an organic osmolyte,compatible solute, antioxidant, or thermal protectant[1,16,17]. Despite the physiological importance of mannitol in brown algae, genes involved in mannitol biosynthesis have been characterized only in Ectocarpus but not in kelps [18],and only a few genes’ functions in this biosynthetic pathway have been confirmed. Moreover, the regulatory mechanisms are yet to be understood.
Saccharina japonica (Sja) is one of the most important brown macroalgae in the order Laminariales because of its considerably high biomass and economic significance [19].Asian countries have been cultivating the Sja species since early 1950s [20], and presently, its annual commercial production (7.7 million tons) ranks the second highest among all aquacultural species [21]. Moreover, purified compounds derived from Sja, such as mannitol, have been widely used as food supplements, medicines, and chemical materials for chemical industry and scientific research [22–25]. The Sja life cycle comprises three stages, including the single-cell (meiospore), multicellular filamentous (gametophyte, n), and large parenchymal individual (sporophyte, 2n) stages [19]. The unique heteromorphic alternation of generations of Sja makes it different from its close relatives in the genus Ectocarpus that lack the parenchymal stage[26].Brown algae are the only secondary endosymbiotic taxa in which multicellularity has evolved [26–28], but the regulatory mechanisms responsible for the structural difference between filamentous brown algae(Ectocarpus) and heteromorphic haploid-diploid algae (Saccharina)are not yet well understood.Moreover,Laminariales,such as Sja,are predominant in the marine ecosystems of cold and temperate coastal zones with harsh and extreme climates[29].
The availability of the Ectocarpus siliculosus genome has paved the way for studying the molecular basis of mannitol biosynthesis in algae [30]. The biosynthesis involves two enzymatic steps: the first step is the reduction of fructose-6-phosphate (F6P) to mannitol-1-phosphate (M1P) by mannitol-1-P dehydrogenase (M1PDH; EC 1.1.1.17), and the second step is the hydrolysis of M1P to mannitol by mannitol-1-phosphatase (M1Pase; EC 3.1.3.22) [1]. A recent analysis of mannitol biosynthesis revealed mannitolmetabolism-associated (MMA) genes in algae. For example,MMA genes encoding M1PDHs and haloacid dehalogenases(HAD-M1Pases) have been found in the members of Phaeophyceae, including Ectocarpales and Laminariales [17,31,32].Previous phylogenetic analyses suggested that these genes were imported into brown algae by horizontal gene transfer from Actinobacteria [18]. Subsequently, a more comprehensive assessment across various algal lineages confirmed that these genes may be present in nonphotosynthetic eukaryotic host cells involved in endosymbiosis [17]. Native M1PDH and M1Pase activities have previously been characterized in cellfree extracts from red algae Dixoniella grisea [6], Caloglossa continua [33,34], and Caloglossa leprieurii [5], brown algae Spatoglossum pacificum, Dictyota dichotoma, and Laminaria digitata [35,36]), and green alga Platymonas subcordiformis[37]; however, genes encoding these enzymes have not been identified.In the model brown alga E.siliculosus,recombinant EsM1PDH1cat (containing only the catalytic domain) and EsM1Pase2 have been characterized [2,32,38]. Determination of kinetic parameters indicated that EsM1PDH1cat exhibited higher catalytic efficiency for F6P reduction than M1P oxidation;EsM1Pase2 was shown to hydrolyze the phosphate group from M1P to produce mannitol, but it was inactive on hexose monophosphates such as glucose-1-phosphate (G1P), glucose-6-phosphate(G6P),and F6P[2,32].Moreover,gene expression analysis showed that transcription of three M1PDHs and two M1Pases in E.siliculosus(a filamentous brown alga)was influenced by the diurnal cycle, and EsM1Pase1 was highly downregulated under hyposaline stress [32]. However, these genes remain poorly illustrated in Sja (a large parenchymal brown alga).
In this study, we systematically characterized the M1PDH and M1Pase genes and their encoded proteins, which are known to act in the Sja mannitol biosynthesis, based on transcriptomic and proteomic data generated from different Sja tissues of various generations (including the sporophyte and gametophyte generations)or under different abiotic stress conditions. Our results extend the understanding of mannitol metabolic pathways and their regulatory mechanisms in the context of ecophysiological and evolutionary significance of Laminariales. In addition, Sja MMA genes may be used to engineer microbes for mannitol production, and also to engineer plants for increasing their tolerance to abiotic stresses and for mannitol biosynthesis for subsequent extraction.
Results
A combined analysis of RNA-seq and Tandem Mass Tag
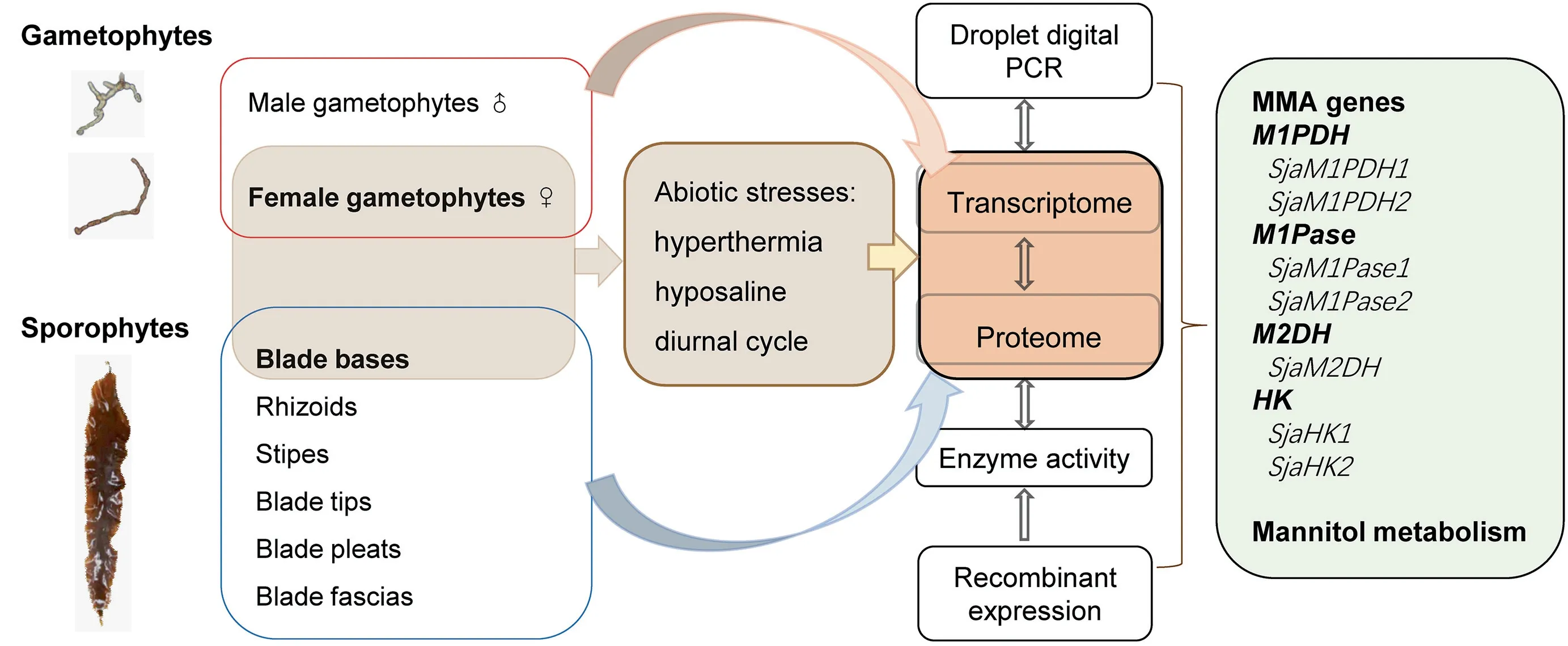
Figure 1 A flow chart of the study designTwelve Saccharina japonica (Sja) samples were collected for transcriptome sequencing, including two separately from male and female gametophytes,six from different parts of thalli(rhizoids,stipes,blade tips,blade pleats,blade bases,and blade fascias),and four from Sja incubated under different abiotic stresses(hyperthermia and hyposaline).Four samples from female gametophytes incubated under different conditions(i.e.,control,hyperthermia,hyposaline,and darkness)were collected for proteome sequencing.Droplet digital PCR(ddPCR)was used to confirm the RNA-seq results.All mannitol-metabolism-associated(MMA)genes from Sja were annotated based on transcriptomic and proteomic data.Some native MMA genes were over-expressed in Escherichia coli to characterize their enzyme activities.
Our combined analysis started with transcriptome sequencing of 12 Sja samples,including two samples separately from male and female gametophytes, six from different parts of thalli(rhizoids, stipes, blade tips, blade pleats, blade bases, and blade fascias), and four from Sja incubated under different abiotic stresses(hyperthermia and hyposaline conditions)(Figure 1). Approximately 9 Gb per algal sample and a total of 336 Gb of RNA-seq raw data were acquired (Table 1). After removing low-quality (< Q20) reads and trimming, the clean reads ranged between 95.21% and 96.70% of the raw reads,and the GC contents ranged from 53.79% to 57.11%. Our Tandem Mass Tag(TMT)proteomic analysis identified a total of 3106 proteins, using samples from female gametophytes incubated under different conditions (i.e., control, hyperthermia,hyposaline,and darkness).The molecular weight of most identified proteins ranged from 20 kDa to 70 kDa (71%, Figure S1A).The sequencing reads with its length 5%longer than its encoded protein length covered 93%(2900/3106)of all analyzed peptides (Figure S1B). About 93% (2888/3106) proteins were inferred from at least two unique peptides (Figure S1C).
MMA genes of brown algae
Combined with our Sja data,we annotated all MMA genes in 19 brown algal species belonging to Laminariales, Ectocarpales, Desmarestiales, Dictyotales, Fucales, and Ishigeales(Table S1). Two unigenes (named M1PDH1 and M1PDH2 according to the naming convention of E.siliculosus M1PDHs)were found in most species; however, the third M1PDH gene(i.e., M1PDH3) was only detected in three species belonging to Ectocarpales (Table S1). The identity between M1PDH1 and M1PDH2 within species appeared low, ranging from 55.10% to 58.59%. The M1Pase genes showed a high degree of conservation in brown algae. Among all 19 species, two M1Pase homologs, named M1Pase1 and M1Pase2, were found and they exhibited intraspecies identities from 55.61%to 68.23% (Table S2). Only one mannitol-2-dehydrogenase(M2DH) gene was found in 15 brown algal species(Table S3), and their identities among the species varied from 69.97% to 96.66%. Two hexokinase (HK) genes, HK1 and HK2, were found in all 19 species, with interspecific identities of 60.98% to 67.43% (Table S4).
Having aligned the brown algal M1PDH amino acid sequences, we identified the conserved blocks A–E of the polyol-specificlong-chaindehydrogenases/reductases(PSLDRs) [39]; M1PDH1 and M1PDH3 showed extended N-terminal domains compared to M1PDH2 (Figure S2A).An alignment of the brown algal M1Pase amino acid sequences with the HAD-like protein ATU0790 from Agrobacterium tumefaciens confirmed the shared catalytic machinery,including motifs I–IV and the Mg2+-cofactor-binding site(Figure S2B).
Function confirmation of the MMA genes
Weoverexpressedcodon-optimizedSjaM1PDH1,SjaM1PDH2, SjaM1Pase1, and SjaM1Pase2 in Escherichia coli to characterize their enzymatic activities.Although several attempts (including different vectors, expression cells, and induction conditions)were made to overexpress SjaM1PDH1,the efforts had failed.Then,the specificity of SjaM1PDH2 was determined by assaying its activity in the presence of different potential substrates, including F6P, G6P, and G1P for detecting reduction activity and M1P, F6P, G6P, and G1P for detecting oxidation activity. First, SjaM1PDH2 had only reduction activity in the mannitol synthesis pathway but not oxidation activity(Table 2).In addition,the reduction activity was detected for other sugar substrates (Table 3), indicating that SjaM1PDH2 is not specific for F6P. Second, purified SjaM1PDH2 had a specific activity of 0.36 μM/min/mg protein for F6P reduction with NADH at pH 8.0. This activity was in the range of those measured for algal M1PDHs, which are listed in Table 2. Using NADPH as an alternative co-factor,the F6P reduction activity of SjaM1PDH2 was less than 20% of that determined in the presence of NADH. Third,the phosphatase activity of SjaM1Pases was determined in 100 mM Tris-HCl buffer, and the specific activity of SjaM1-Pase1(144.93 μM/min/mg protein)was significantly higher(almost 22 folds) than that of SjaM1Pase2 (6.60 μM/min/mg protein) in the presence of 1 mM M1P (Table 4). Forth, the phosphatase activity of SjaM1Pases was also detected for F6P,G1P,and G6P.The activity of SjaM1Pase1 for such substrates was always lower than that for M1P, as observed for most M1Pases from brown and red algae (Table 5). However,SjaM1Pase2 exhibited the highest phosphatase activity in the presence of G1P, similar to the M1Pase from D. grisea showing the highest phosphatase activity using G1P (Table 5). In addition, more than 90% of the enzymatic activities were detected for SjaM1PDH2 and both SjaM1Pases after storage at 4 °C for 72 h, suggesting that the recombinant proteins are quite stable under the purification conditions.
We also evaluated the enzyme activity of the three recombinants under different temperatures and pH conditions. As shown in Figure 2A, the optimal temperature for SjaM1PDH2 was 40 °C, whereas the enzyme activities were 82% and 91% of the maximum at 30 °C and 50 °C, respectively. The optimal temperature for SjaM1Pase1 was 50 °C,with 95% and 83% residual activities at 40 °C and 60 °C,respectively. The optimal temperature for SjaM1Pase2 was 30 °C, which was much lower than that of SjaM1Pase1,with less than 54% residual activities at other temperatures.As shown in Figure 2B, the optimal pH for SjaM1PDH2 was 8.0, with 51% and 56% residual activities at pH 7.0 and 9.0, respectively. The optimal pH of SjaM1Pase1 and SjaM1Pase2 was the same: 8.5.
The recombinant SjaM1PDH2 and SjaM1Pases exhibited typical Michaelis-Menten kinetics when assayed in increasingconcentrations of their substrates, and the apparent Vmaxand Kmvalues were determined based on Lineweaver-Burk plots(Figure 2C). The Kmvalue for SjaM1PDH2 was 2.02 mM for F6P reduction with NADH, which was approximately 10-fold higher than that for EsM1PDH1cat (0.19 mM), indicating lower substrate-binding capacity than that of EsM1PDH1cat (Table 2). The M1P substrate-binding capacities of the two SjaM1Pases were very different: the binding capacity of SjaM1Pase2 (Km= 0.02 mM) was about 41-fold higher than that of SjaM1Pase1 (Km= 0.83 mM), and it was the highest(by 21–315 folds)among the binding capacities of M1Pases from brown and red algae. Interestingly, both SjaM1Pases showed much higher catalytic efficiencies and catalytic rates than EsM1Pase2 (Table 4); SjaM1Pase1 has the highest kcatvalue(almost four orders of magnitude higher than that of EsM1Pase2).
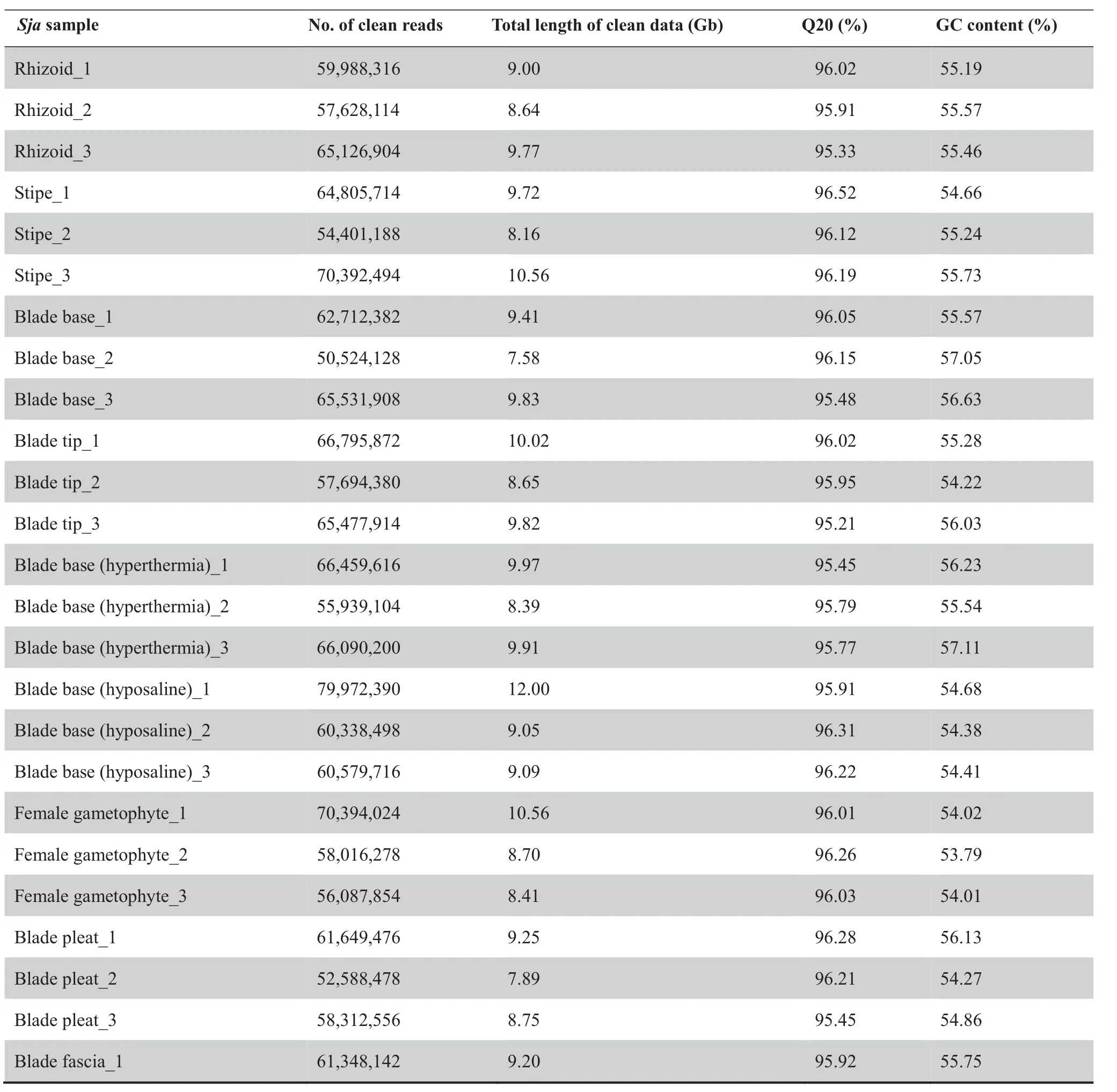
Table 1 Summary of RNA-seq data from all samples
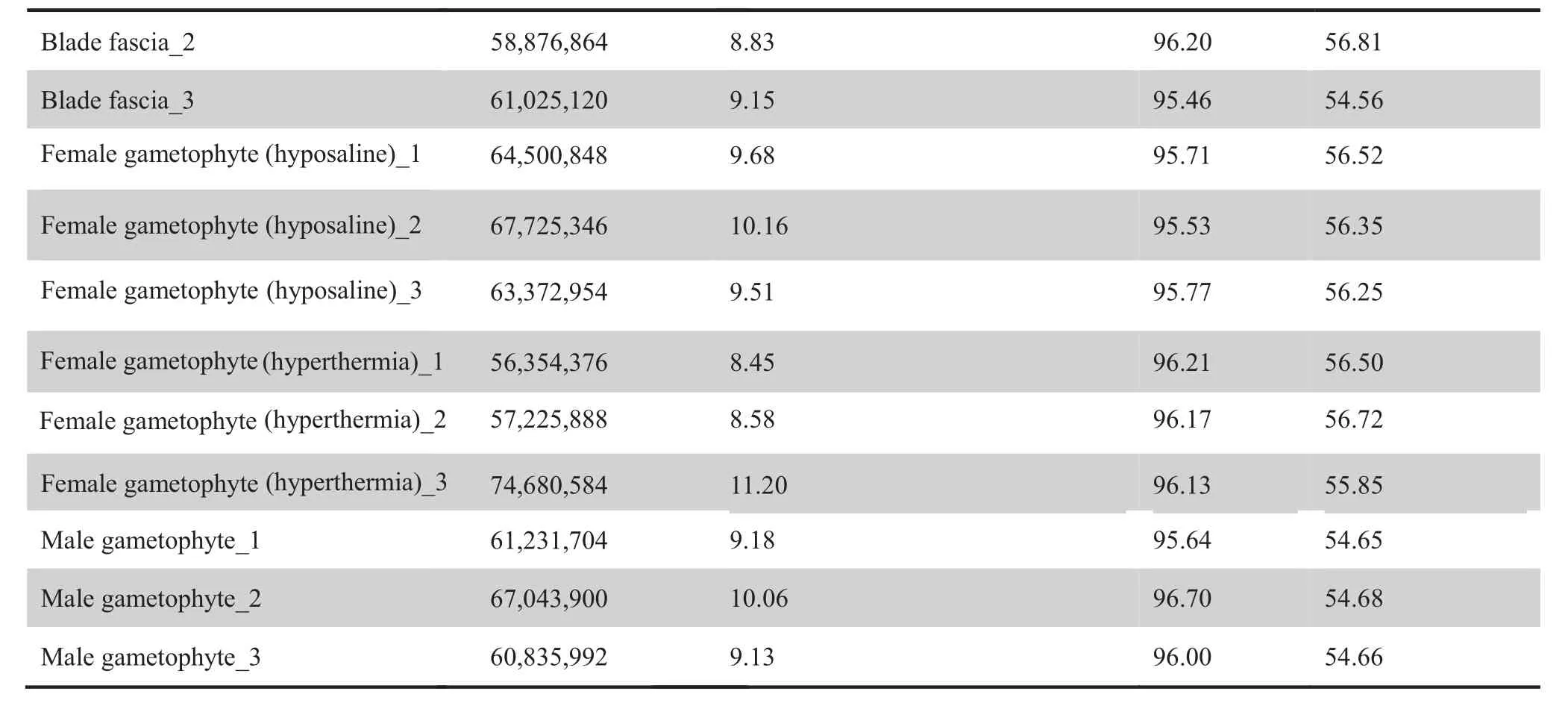
Table 1 (continued)

Table 2 Characterization of M1PDHs in brown and red algae
We then assayed the salt influence on the recombinant SjaM1PDH2 and SjaM1Pases using NaCl (Figure 2D). The results showed that SjaM1PDH2 activity altered under different NaCl conditions: it increased in the presence of 200 mM NaCl but decreased dramatically when NaCl concentration changed to 400 mM, showing a near-linear relationship from 400 mM to 1000 mM. For SjaM1Pase1 and SjaM1Pase2, a nearly linear decrease in activity was observed in the presence of NaCl at concentrations ranging from 0 mM to 1000 mM.Approximately 60% of the initial activities of both SjaM1-Pases remain in the presence of 1000 mM NaCl, suggesting that these SjaM1Pases may be salt-resistant.
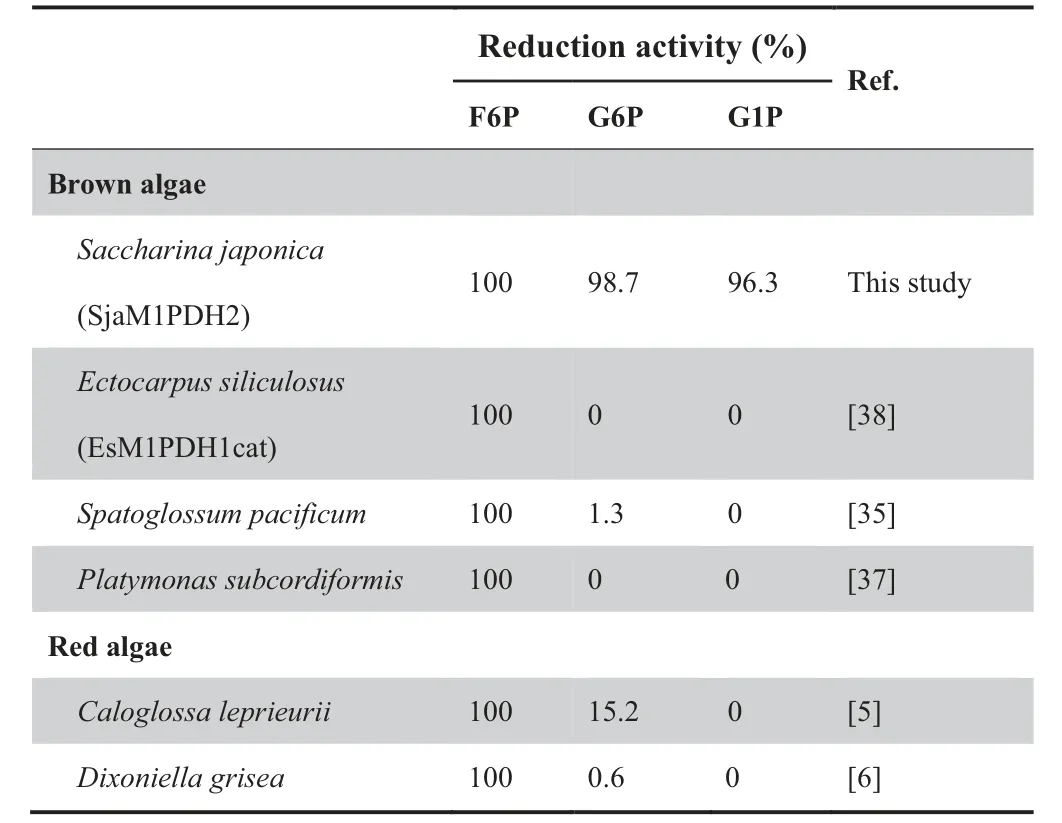
Table 3 M1PDH reduction activity determined in brown and red algae
Expression of the MMA genes
Our Sja MMA gene expression study relies on both transcriptomic and proteomic data (db.cngb.org/onekp/). All of the 19 Phaeophyceae species appear to express two M1Pase genes and two HK genes (Tables S2 and S4). Nearly 80% of the 19 Phaeophyceae species appear to express two M1PDH genes and one M2DH genes at the transcriptional level, but these genes were not identified in the remaining 20% of the Phaeophyceae species assessed in this study because of low coverage or quality of some of the transcriptomes(Tables S1 and S3).At both the transcriptomic and proteomic levels, all seven Sja MMA genes were detected (Tables S5 and S6), and droplet digital PCR (ddPCR) was also performed to confirm our results (Table S7).
The regulation of Sja mannitol metabolism is highly complex, and four observations have been made in general. First,these MMA genes (including different gene family members)were all expressed constitutively. As shown in Figure 3, all seven MMA genes (representing four enzymes) were detected in all samples. The range of fragments per kilobase of transcript per million mapped reads (FPKM) values was 1.2–300(Table S5).
Second, most MMA genes encode reversible enzymes (except SjaM1PDH2 and two SjaM1Pases), which control the balance between mannitol and F6P and dynamically maintain the ‘‘carbohydrate pool” in vivo. Mannitol metabolism is closely associated with the metabolism of alginate,fucoidan,laminarin, and trehalose via the intermediate product F6P. The first gene that transforms F6P to mannitol is M1PDH(M1PDH1 and M1PDH2), and the first gene that transforms F6P to alginate and fucoidan is mannose phosphate isomerase MPI (MPI1 and MPI2) [40]. The expression of two SjaM1PDH genes as well as two SjaMPI genes was compared among different tissues (Figure 4A and B). For example, the expression of SjaMPI1 was 3.8-fold higher than that of SjaM1PDH1 in blade fascias, whereas the expression of SjaM1PDH1 is 2.7-fold higher than that of SjaMPI1 in stipes.
Third, expression of most MMA genes differed among different tissues and under various stresses (Figures 3 and 4).Regarding to the two M1Pase family members,the expression of SjaM1Pase1 was 2.0–19.4-fold higher than that of SjaM1-Pase2 in different tissues (Figure 4C). However, under hyperthermia stress (18 °C), the expression of SjaM1Pase2 was clearly up-regulated by a 2-fold increase, whereas the expression of SjaM1Pase1 is down-regulated by a 3-fold decrease.
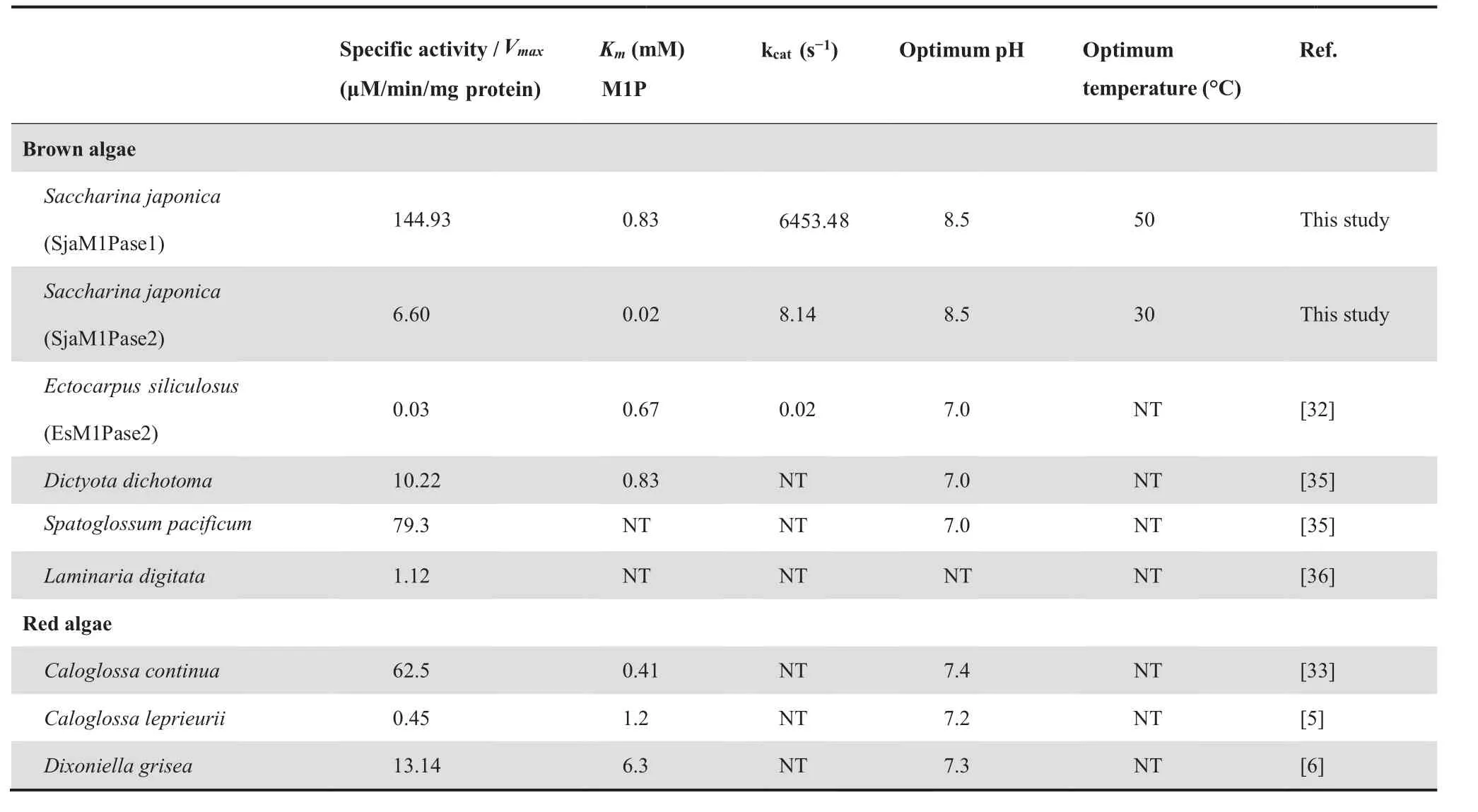
Table 4 Characterization of M1Pases in brown and red algae
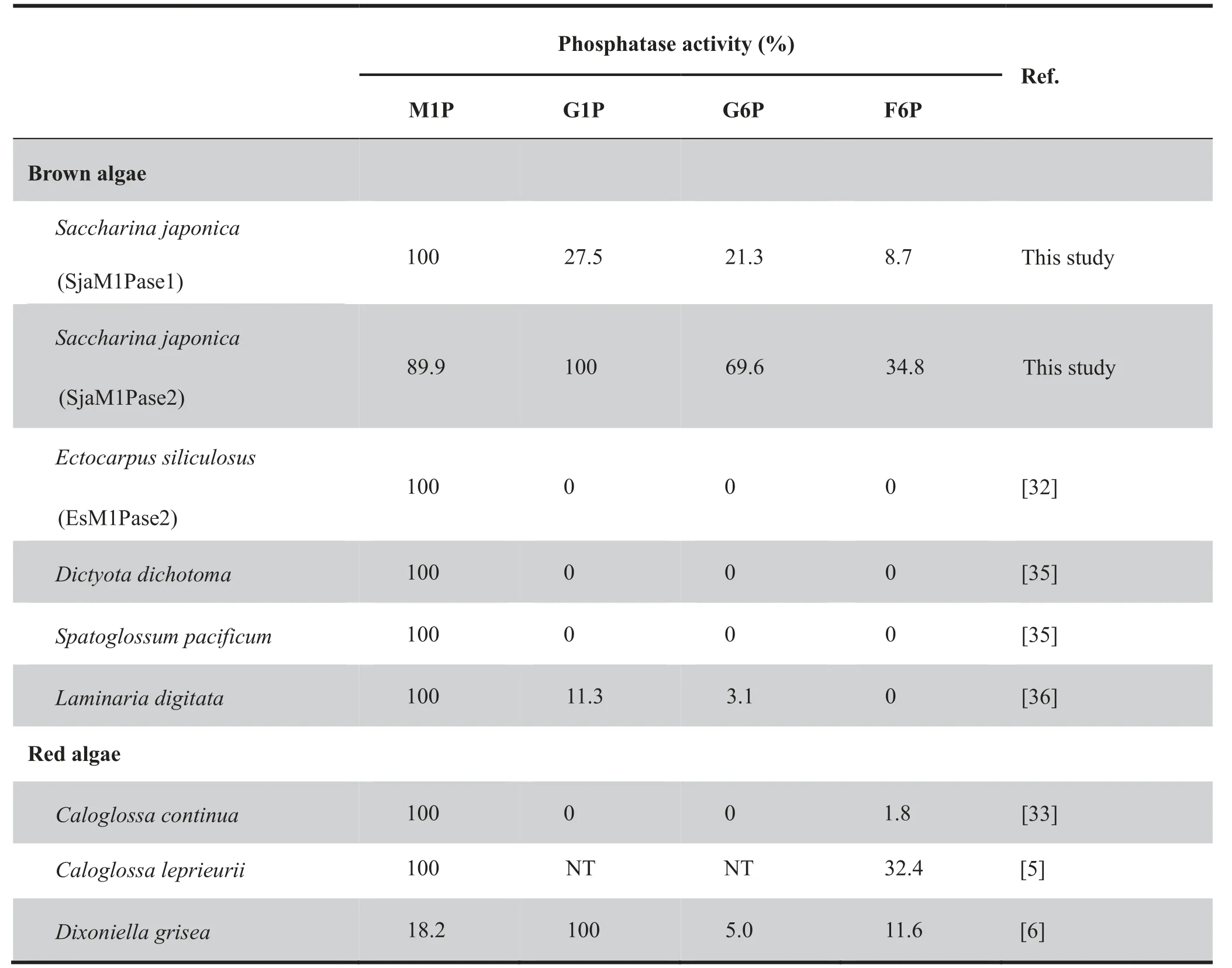
Table 5 M1Pase activity determined in brown and red algae
Finally, the overall gene expression profiles differed between the gametophyte and sporophyte generations. Most MMA genes(SjaM1PDH1,SjaM1Pase1,SjaM2DH,SjaHK1,and SjaHK2) were expressed at significantly higher levels in sporophytes than in gametophytes, showing 2.8-, 7.3-, 3.1-,5.6-, and 4.7-fold changes, respectively (Figure 5A). However,the expression levels of SjaM1PDH2 and SjaM1Pase2 did not differ significantly as compared between the two generations.In addition, there was no significant expression difference among all MMA genes between the female and male gametophytes (Figure 5B).
We further investigated MMA gene expression under abiotic stresses. Under hyposaline conditions, the transcriptional levels of all MMA genes were elevated, exhibiting increases of 1.2–16.2 folds in gametophytes. In contrast, the expression levels of all MMA genes decreased in 1.9–3.7 folds in sporophytes (Table 6). Furthermore, these changes followed a similar trend under hyperthermia conditions:at the transcriptional levels, all MMA genes were up-regulated (1.2–12.5 folds) in gametophytes, whereas most of them (except SjaM1PDH2 and SjaM1Pase2) were down-regulated (1.4–3.0 folds) in sporophytes (Table 6).
Discussion
Constitutively expressed MMA genes satisfy the requirement for mannitol biosynthesis and accumulation
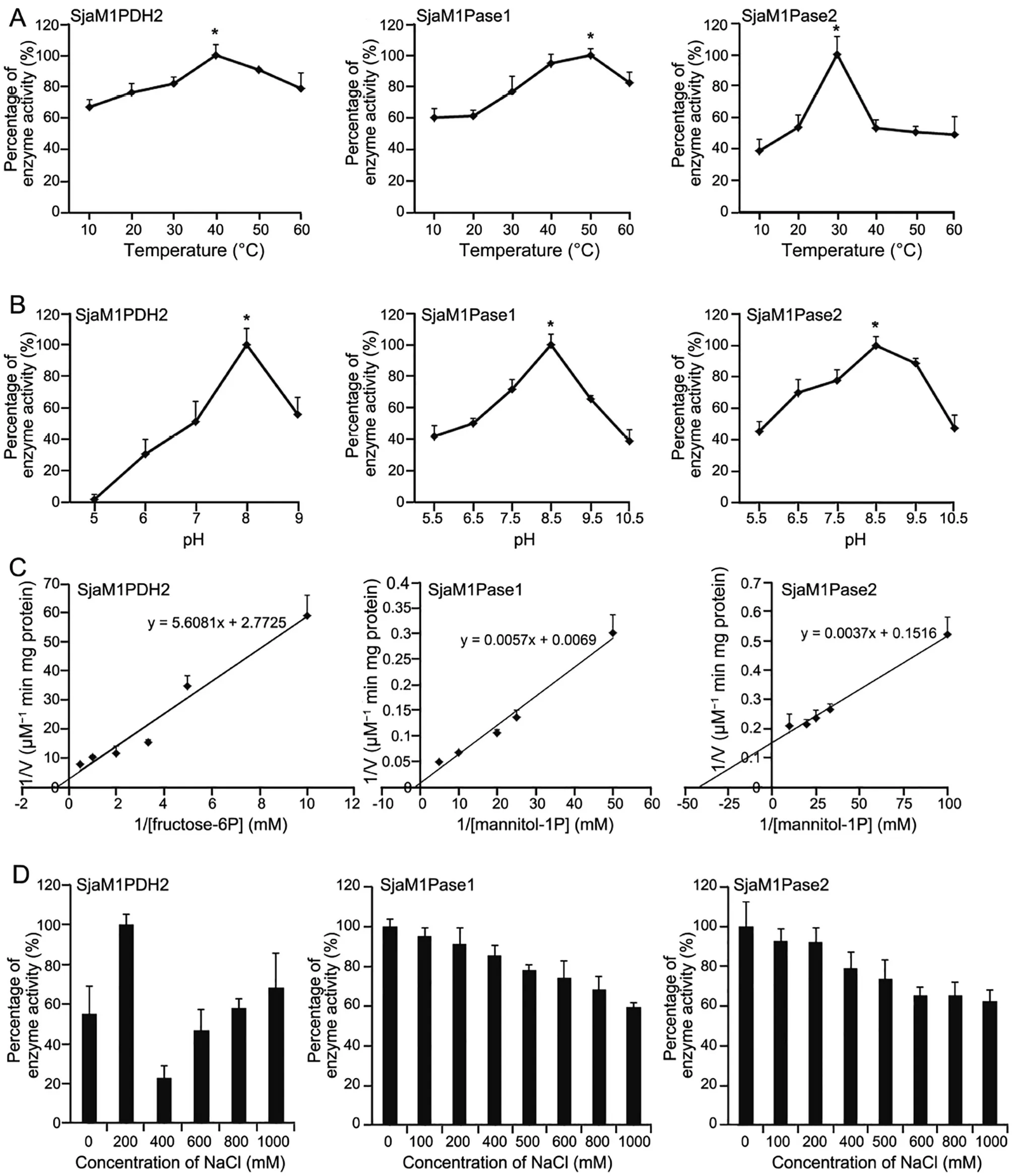
Figure 2 Enzymatic characterization of recombinant SjaM1PDH2, SjaM1Pase1, and SjaM1Pase2A. Temperature influence on enzyme activities of SjaM1PDH2, SjaM1Pase1, and SjaM1Pase2. The activities at 40 °C, 50 °C, and 30 °C are set to be 100% for SjaM1PDH2, SjaM1Pase1, and SjaM1Pase2, respectively. B. pH influence on enzyme activities of SjaM1PDH2,SjaM1Pase1,and SjaM1Pase2.The activities at pH 8.0,8.5,and 8.5 are set to be 100%for SjaM1PDH2,SjaM1Pase1,and SjaM1Pase2,respectively.C.Kinetics of enzyme activities of SjaM1PDH2,SjaM1Pase1, and SjaM1Pase2. D.The influence of NaCl concentration on enzyme activities of SjaM1PDH2, SjaM1Pase1, and SjaM1Pase2. The values represent mean ± SD which are calculated from three replicates.
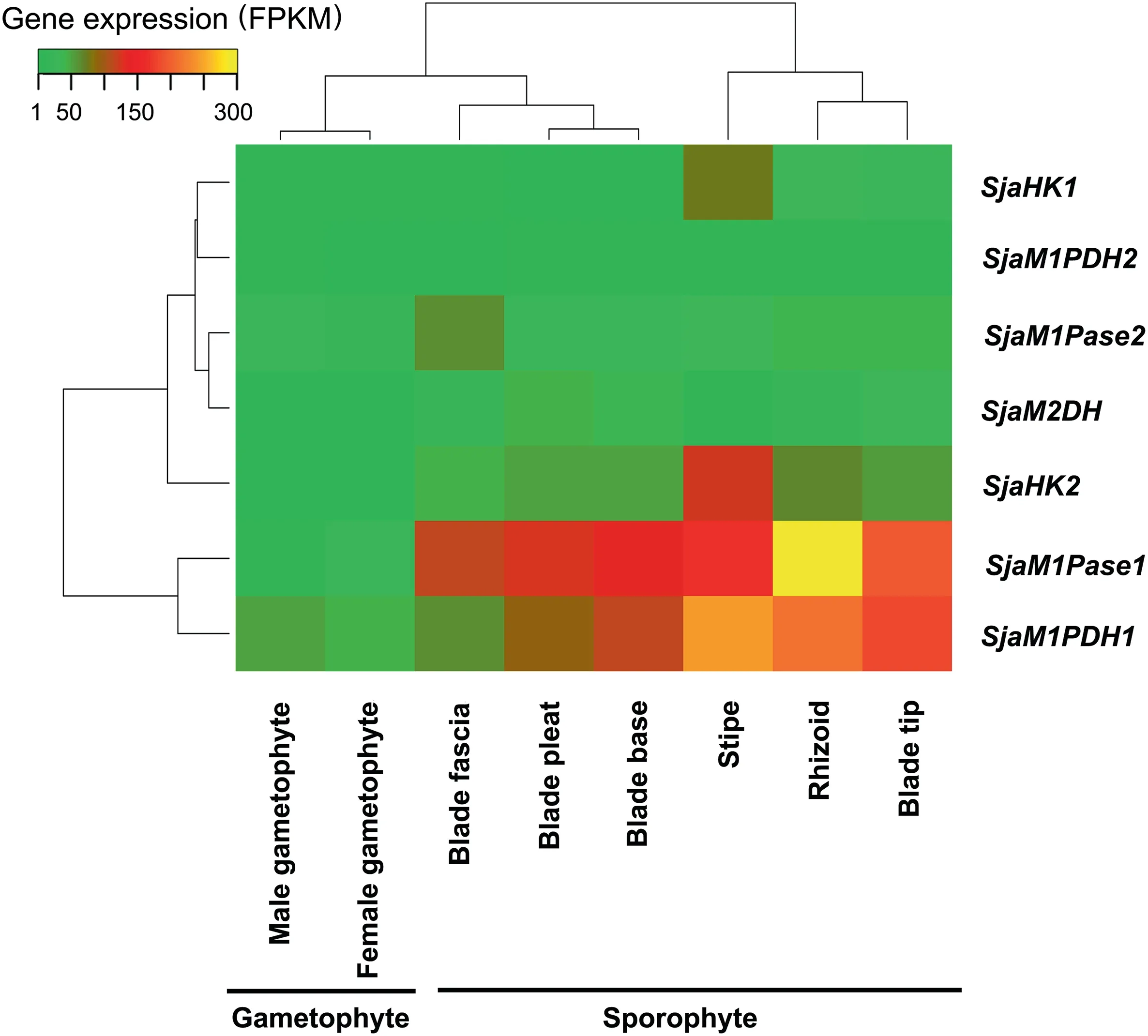
Figure 3 Expression of Sja MMA genes in different generations and tissuesAll seven Sja MMA genes (representing four enzyme families) were constitutively expressed in various generations (gametophytes and sporophytes)and different tissues(rhizoids,stipes,blade tips,blade pleats,blade bases,and blade fascias).The fragments per kilobase of transcript per million mapped reads(FPKM)of each gene was calculated based on the length of the gene and the number of reads mapped to the gene. The results represent mean values of three replicates.
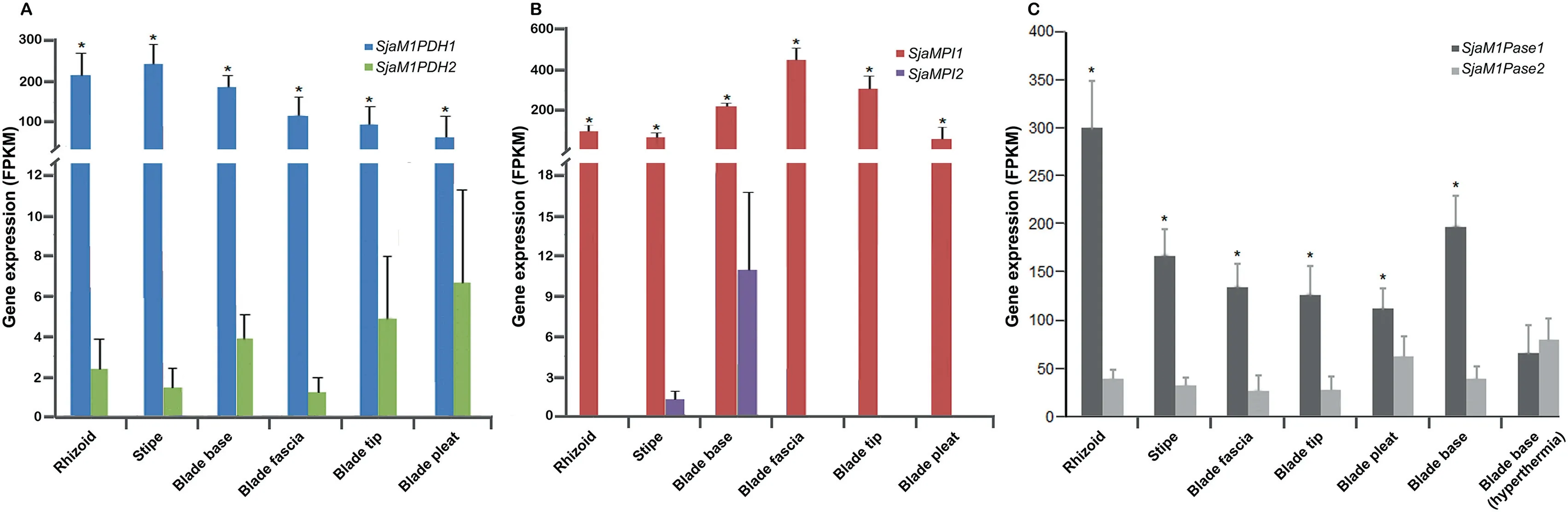
Figure 4 Expression of Sja MMA genes in different tissues and under hyperthermia stressA. The expression of SjaM1PDH1 was significantly higher than that of SjaM1PDH2 in all tissues. B. The expression of SjaMPI1 was significantly higher than that of SjaMPI2 in all tissues.C.The expression of SjaM1Pase1 was significantly higher than that of SjaM1Pase2 in all tissues. The expression levels of SjaM1Pase1 and SjaM1Pase2 were affected by hyperthermia stress (18 °C). All the data are subjected to one-way analysis of variance followed by Student’s t-tests. *, P < 0.05.
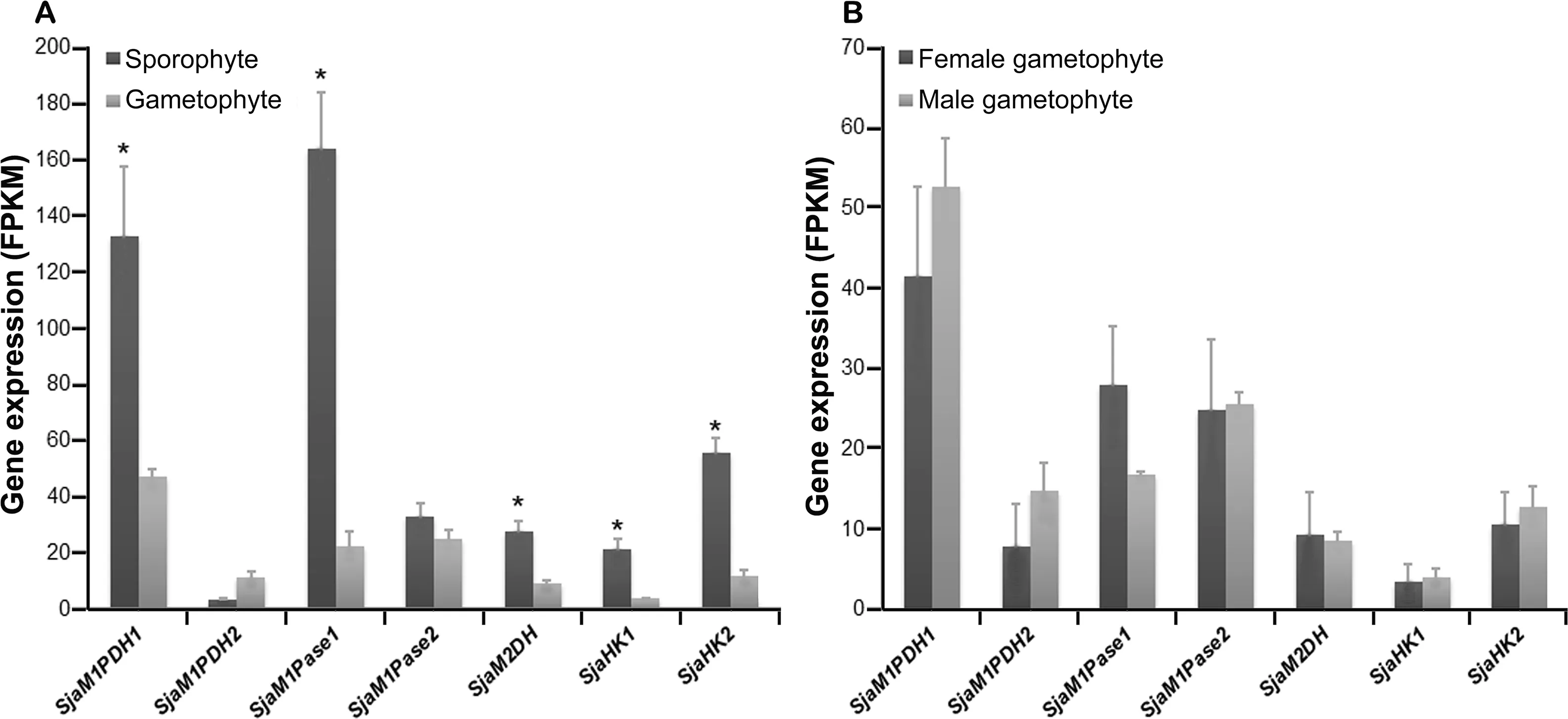
Figure 5 Expression of Sja MMA genes of different generations and sexesA. Most Sja MMA genes in sporophytes were expressed significantly higher than those in gametophytes. B. No significant expression differences were observed between sexes. All the data are subjected to one-way analysis of variance followed by Student’s t-tests. *,P < 0.05.
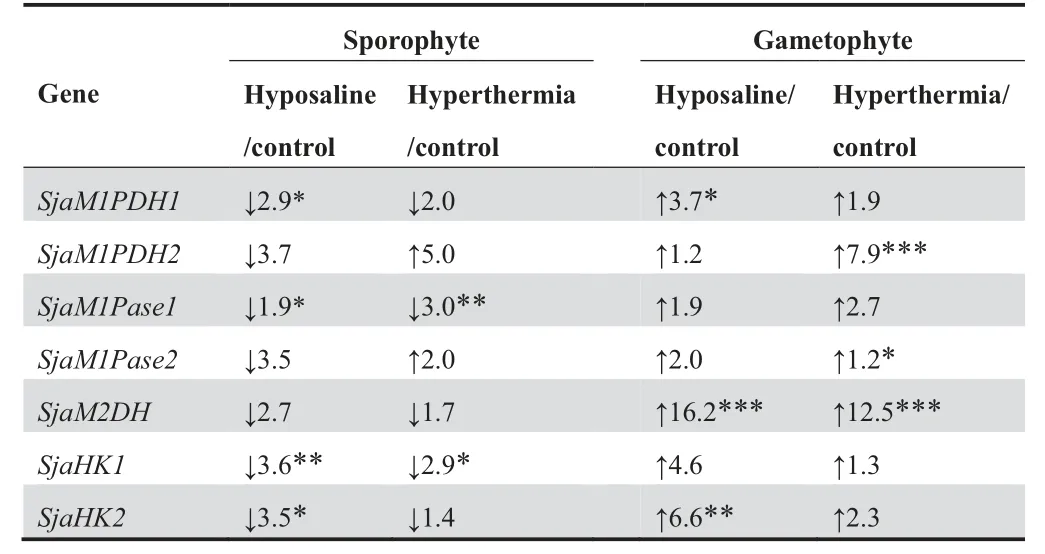
Table 6 Ratios of FPKM values of MMA genes under different conditions
Mannitol is one of the fundamental carbon-storage molecules and osmotic regulators in brown algae,and mannitol metabolism is one of the main traits that make brown algae unique as compared to other eukaryotic algae [41]. As a key metabolic pathway, mannitol metabolism has a total of four steps, and involves a limited number of gene family members (1–2 members of each gene family in most brown algae),as compared to,for instance, the Sja halogen metabolism (unpublished data)that has approximately 89 gene family members. As a singleproduct pathway, the mannitol metabolism does not contain complex synthesis- and modification-associated genes, such as those encoding glycosyltransferases, sulfurtransferases,and mannuronate C5-epimerases, as observed in alginate and fucoidan metabolism that contains dozens to more than 100 genes [40]. Therefore, mannitol metabolism is rather unique in studying pathway regulations and environmental adaptations in brown algae.
There are three M1PDH unigenes in E.siliculosus,but only two unigenes are present in the Sja genome. Our transcriptomic data of 19 Phaeophyceae species enable the identification of the two M1PDH unigenes expressed in most brown algae samples (Table S1). The presence of a third unigene of M1PDH in Ectocarpales may be explained by gene duplication
[17].With regard to the M1Pase family members,unlike other primary endosymbiotic (e.g., red algae) and secondary endosymbiotic (e.g., Dictyochophyceae from Ochrophyta)algae,which have only one copy of this gene[17],brown algae have two HAD-M1Pase genes (Table S2). In addition, our results show that all seven MMA genes are constitutively expressed in various Sja generations and among different Sja tissues (Figure 3; Table S5). In our proteomic study, proteins encoded by these MMA genes are all expressed in Sja female gametophytes (Table S6). Interestingly, no significant difference is observed under hyposaline and hyperthermia stresses at the proteomic level. Although up-regulation at the transcriptional level under stresses is shown as a stable trend(Table S7), the increase in gene expression doesn’t necessarily imply an increased production of corresponding proteins.Even under dark conditions, all proteins involved in the pathway remain constant in Sja female gametophytes during diurnal cycle (Table S6).
During Sja development, zygotes divide continuously from a single cell to form thallus sporophytes, exhibiting consistent increases in length, width, and thickness. Mannitol of brown algae is a central compound in carbon metabolism and in the transportation and distribution of the organic assimilate.Moreover, mannitol has important physiological functions,such as osmotic regulation, thermal protection, or acting as an antioxidant and respiratory substrate [42,43]. Therefore,these results suggest that brown algae consistently synthesize mannitol for carbon and energy storage throughout their life cycle.
Specific MMA gene regulation in the Sja mannitol cycle
As a shared substrate, F6P can be used to synthesize not only mannitol but also other cellular components (e.g., alginate and fucoidan), and MMA genes in Laminariales may be regulated in a complex integrated system. For example, the expression levels of the first genes in the mannitol and alginate/fucoidan metabolic pathways differ substantially among tissues(Figure 4A and B),indicating that abundant F6P could be utilized for the synthesis of alginate and fucoidan. This result is consistent with the finding that the accumulation of mannitol has an inverse relationship with that of alginate and fucoidan [44]. The balance between mannitol and F6P can be stringently regulated in mannitol metabolism pathway,which further affects other related pathways, such as alginate and fucoidan metabolism. It is beneficial for the maintenance of a viable carbohydrate pool in vivo.
M1Pase1 is a key MMA gene in mannitol synthesis
M1Pase catalyzes an irreversible reaction of the mannitol biosynthetic pathway, which is also a rate-limiting step.Although the two M1Pase genes appear to be expressed constitutively,they have substantial differences in expression patterns.M1Pases exhibit mannitol biosynthetic activity and have different biochemical properties.In brown algae,only one homolog of M1Pases (i.e.,M1Pase2) has been confirmed to be active in E.siliculosus[32,38].No enzymatic studies have been conducted for MIPase1;only the nucleotide sequence of MIPase1 from E.siliculosus has been reported. In this study, SjaM1Pase1 and SjaM1Pase2 are both confirmed to have M1Pase activity and assumed to be involved in mannitol biosynthesis in brown algae(Figure 2).The specific enzyme activity of SjaM1Pase1 is much higher (22.0 folds) than that of SjaM1Pase2, and the former enzyme has a higher kcatvalue (792.8 folds) than the latter(Table 4).This result indicates that SjaM1Pase1 has a higher catalytic activity than SjaM1Pase2. SjaM1Pase1 also has significantly higher expression levels than SjaM1Pase2 in all samples(Figure 4C),suggesting that SjaM1Pase1 is the key MMA gene for mannitol production in Sja,whereas SjaM1Pase2 may play complementary roles under other defined conditions.For example, while the expression of SjaM1Pase1 is down-regulated under hyperthermia stress in sporophytes,SjaM1Pase2 exhibits an opposite trend under the same temperature stress.
Gene duplication in evolution is an important general mechanism for maintaining normal biochemical metabolism under variable conditions,and it is probably in action here for the photosynthetic carbon storage in Sja. Different copies of MMA genes are observed to be transcribed,and their products exhibit activities that prevent gene loss, recombination, and mutation from affecting the pathway integrity and adequate enzymatic function. Interestingly, most genes are down-regulated under hyperthermia stress in sporophytes,whereas SjaM1PDH2 and SjaM1Pase2 are up-regulated(Figure 4C;Table S5).These findings suggest that paralogs encoding proteins with the same enzyme activity may be regulated by different mechanisms and/or under difference environmental conditions.In addition,SjaM1PDH2 lacks M1P oxidation activity. A previous study has shown that EsM1PDH1cat can oxidize M1P [38]. In our analysis, SjaM1PDH1 and EsM1PDH1cat have highly conserved domains(Blocks A–E,Figure S2),and we thus speculate that the M1P oxidation activity may be conducted by SjaM1PDH1 in Sja, whereas SjaM1PDH2 only maintains the reduction activity. However, the reduction activity of SjaM1PDH2 is not specific to F6P, suggesting that it may be involved in other metabolic pathway(s),such as phosphoglucose reduction.This functional differentiation between homologous genes is similar to the MPI genes in alginate/fucoidan metabolism[40].
The complex regulation of MMA genes is beneficial for brown algal evolution and adaptation
Laminariales have a heteromorphic haploid-diploid life cycle,with a macroscopic thallus sporophyte and a microscopic gametophyte among different generations [19]. Most MMA genes (except SjaM1PDH2 and SjaM1Pase2) exhibit significantly higher expression in sporophytes (thallus generation)than in gametophytes (filamentous generation) (Figure 5A).These results indicate that sporophytes may have a much greater ability to synthesize mannitol than gametophytes,potentially because sporophytes (large thallus with tissue differentiation) require high levels of mannitol not only for normal growth and development, but also for adaptation to changing environments. However, there are no significant differences in gene expression between female and male gametophytes (Figure 5B). This result is consistent with our observation that the mannitol contents in male and female gametophytes are very similar in quantity, 23.4% and 24.6%(P > 0.05), respectively (unpublished data).
The expression levels of MMA genes in response to environmental stresses show opposite trends between gametophyte and sporophyte generations. Under hyposaline stress, the expression of all MMA genes in gametophytes is upregulated, consistent with the results observed for M1Pase2 in E. siliculosus [32]; on the contrary, the expression of all MMA genes in sporophytes is down-regulated (Table 6). Similar results are observed under hyperthermia condition. Notably, mannitol is the main organic osmolyte in Sja and most other brown algae, counteracting salinity stress and acting as an antioxidant and heat protectant for protein stabilization[1,42,43].The gametophyte stage is most vulnerable to external stresses in the entire life cycle [45]. Therefore, in this stage,mannitol metabolism is increased in order to respond to stresses,particularly hyposaline.Distinct regions of carbon sources and carbon sinks exist along thalli because of their large size and morphological differentiation [46,47]. Saccharina species are capable of transporting carbons from source(mature blade areas) to sink, producing a surplus of photoassimilates transported to intercalary carbon-requiring meristems [19]. The imported organic compounds in the sink tissues are rapidly metabolized and incorporated into polysaccharides and proteins[46].The blade bases of the sporophytes(meristem)exhibit reduced mannitol catabolism, indicating a reduction of mannitol degradation and incorporation [48]. We speculate that this condition may decrease transportation from carbon sources to carbon sinks and increase mannitol accumulation in mature blades for stress responses.
The mutual validation of RNA-seq and ddPCR results provides confidence in copy number quantitation
We performed ddPCR, a microfluidics-based technique for determination of DNA or RNA copy number in a given sample,to verify our key RNA-seq results in female gametophytes under stress conditions (hyposaline and hyperthermia). Most of our gene expression results are confirmed. For example, in RNA-seq analysis, SjaM1PDH1 expression shows 3.75- and 1.87-fold increases under hyposaline and hyperthermia stresses, respectively, and the ddPCR analysis accordingly shows 3.97-and 1.30-fold increases,respectively(Table S7).Notably,ddPCR is relatively inexpensive and has shorter experimental turnaround time of RNA-seq.
Characterization of MMA genes is essential for future industrial production and genetic breeding
Mannitol is a commercially valuable compound widely used in food,pharmaceutical,medical,and chemical industries[23,49].Most of mannitol commercial production is carried out by chemical hydrogenation of fructose or by extraction from seaweed [13,23]. Since environmental issues associate with chemical refine and production, microbe-based alternatives have been a subject of significant interest in recent years [23]. The most widely used M1Pase gene, from the protozoan parasite Eimeria tenella [31], has been expressed in Cyanobacteria[50],Proteobacteria[51],and Firmicutes[52]to generate cellular or extracellular mannitol. Interestingly, the substratebinding capacity of the E. tenella M1Pase (Km= 0.07 mM)is lower than that of SjaM1Pase2 (Km= 0.02 mM), and the catalytic efficiency of the E. tenella M1Pase (kcat= 430 s-1)is much lower than that of SjaM1Pase1 (kcat= 6453.48 s-1),indicating that the M1Pase genes from brown algae may be suitable candidates for engineered microbes for mannitol production. Furthermore, introduction of algal genes into transgenic plants may confer great advantage in terms of salt tolerance [33]. For example, mannitol biosynthesis by genetic engineering is one of the most extensively tested alternatives for improving salinity tolerance in plants[1].The high salinity tolerance of transgenic plants may be a result of both the accumulation of mannitol in cells [53,54] and increased expression of a variety of stress-inducible genes [55]. Analysis of MMA genes in the mannitol synthesis pathway helps identify enzymes with high substrate specificities and activities,which are useful for genetic breeding of both algae and plants in the future.
Materials and methods
Algal sample collection
Preserved Sja haploid gametophytes(male and female gametophytes) were available in our laboratory cultures (Laboratory of Genetics and Breeding of Marine Organisms, Ocean University of China),which were grown in a modified seawater medium supplemented with nutrients (4 mg/L NaNO3-N and 0.4 mg/L KH2PO4-P) at 10 °C under 30 μM photons m-2s-1irradiance. Fresh samples of Sja sporophytes were collected from eastern China (Rongcheng, Shandong Province,37°8′53′′N, 122°34′33′′E) and kept in the laboratory under the same culture conditions as gametophytes for a few days to acclimate,and then dissected to obtain different tissues(rhizoids, stipes, blade tips, blade pleats, blade bases, and blade fascias). To study the effects of abiotic factors, the female gametophytes (5 g each sample) and blade bases of sporophytes (5 g each sample) were cultured at different temperatures (control: 8 °C; hyperthermia: 18 °C) and salinities(control: 30‰; hyposaline: 12‰) for 6 h; the female gametophytes(5 g each sample)were cultured under a 12-h/12-h photoperiod (control: collected at the end of 12-h light period;darkness: collected at the end of 12-h dark period).
Sequence analysis
Based on the analysis of the Sja genome sequence downloaded from Genome Warehouse (GWH: GWHAAET00000000;http://bigd.big.ac.cn/gwh/browse/index), the transcriptome database downloaded from 1000 plant trancriptomes(OneKP:OGZM; https://db.cngb.org/onekp/), and the transcriptome and proteome databases obtained in our study, the unigenes related to mannitol metabolism pathway were verified using the BLASTX algorithm (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Multiple sequence alignments were performed with ClustalX[56].Sequence identities were calculated using the Clustal Omega tool (http://www.ebi.ac.uk/Tools/msa/clustalo/).
Purification of recombinant proteins expressed in E. coli
Genes were codon-optimized and synthesized (Shanghai Xuguan Biotechnological Development Co., Ltd, Shanghai,China) to construct recombinant plasmids. SjaM1PDH1,SjaM1PDH2, and SjaM1Pase1 were cloned into pET32a,and SjaM1Pase2 was cloned into pGEX-6p-1.The inserts were cloned into the plasmids between the EcoRI and NotI sites.After ligation,the generated recombinant plasmids were transformed into E. coli BL21 (DE3) cells, and the integrity of the sequences was verified based on Sanger sequencing.
The recombinant strains were cultivated in 1 L LB liquid medium. When the optical density at 600 nm (OD600) reached 0.6,isopropyl β-D-1-thiogalactopyranoside(IPTG)was added at a final concentration of 0.5 mM to induce overexpression of the target genes to produce recombinant proteins,and the bacterial cultures were incubated for 16 h at 20°C.Then,the cells were harvested by centrifugation at 12,000 g for 3 min at 4°C,suspended in 20 ml of 50 mM Tris-HCl buffer (pH 8.0), and lysed by sonication (Selecta Sonopuls, Shanghai, China) to release the recombinant proteins. His-binding resin and GST-binding resin were used to purify the recombinant proteins according to the manufacturer’s instructions (www.yuekebio.com). The proteins were stored at -80 °C.
Enzyme kinetic assays
The enzyme activities of recombinant SjaM1PDH2, SjaM1-Pase1, and SjaM1Pase2 were determined using previously described methods [32,38]. For enzymatic characterization,four sugar and polyol phosphoesters considered as potential substrates were tested: M1P, F6P, G1P, and G6P (Sigma, St.Louis, MO). The pH effect on the enzyme activity was determined at pH values ranging from 5.0 to 9.0 for SjaM1PDH2 and 5.5 to 10.5 for SjaM1Pases. The temperature effect was determined at temperatures ranging from 10 °C to 60 °C.The NaCl influence was assessed at final concentrations ranging from 0 mM to 1000 mM in the reaction mixture. Four replicates were analyzed for each condition to ensure reproducibility of the experimental results. In each case, the heatdenatured recombinant enzyme was used as a negative control.
RNA-seq and ddPCR
Total RNA was extracted by using an improved CTAB method[57].Three micrograms of total RNA per sample were used for the construction of sequencing libraries.mRNAs were enriched by using oligo-dT beads, and sequencing libraries were generated by using the NEBNext Ultra RNA Library Prep Kit (NEB, Ipswich, MA) for Illumina according to the manufacturer’s instructions. Then, index codes were added to attribute sequences to each sample. Clustering of the index-coded samples was performed on a cBot cluster generation system(TruSeq PE Cluster Kit v3-cBot-HS,Illumina,San Diego, CA) according to the manufacturer’s instructions.After cluster generation, the libraries were sequenced on the Illumina HiSeq platform, and the length of paired-end reads was 2 × 150 bp. High-quality reads (quality above Q20 sequences, no adaptor contamination, and no ambiguous N bases) were aligned to the Sja genome (NCBI accession No.JXRI00000000.1) and to a set of gene model annotations including MMA genes using HISAT2 [58]. HTSeq v0.6.1 was used to count the read numbers mapped to each gene [59].The FPKM of each gene was then calculated based on the length of the gene and the number of reads mapped to the gene.
ddPCR analysis was conducted according to previously described methods[40].Each 20 μl reaction mixture contained:1× Droplet PCR Supermix (Bio-Rad, Hercules, CA), 3 μl of sample cDNA, 900 nM of each primer, and 250 nM of the probe(Table S8).The 20 μl mixture was mixed with 70 μl droplet generation oil in the Droplet Generator (Bio-Rad, Hercules, CA) via microfluidics. The water-in-oil droplets were transferred to a standard 96-well PCR plate for PCR amplification. The PCR program included an initial denaturing step at 95 °C for 10 min, followed by 40 cycles of 94 °C for 30 s and 60 °C for 60 s, and a final step at 98 °C for 10 min. The results represented mean values of three replicates. All data were subjected to one-way analysis of variance followed by Student’s t-test.
Protein identification by LC–MS/MS
Every five samples, including three treatments, a control, and an internal standard (IS; all samples pooled equally), were labeled as a replicate, and three replicates were taken for each measurement. Proteins were extracted using the phenol/SDS method described by Nagai K et al. [60]. Protein digestion was conducted according to the filter-aided sample preparation(FASP) procedure [61]. TMT6plex labeling was conducted according to the manufacturer’s recommendations (Thermo-Fisher Scietific, Waltham, MA). The TMT-labeled peptides were subjected to high-pH reversed-phase fractionation in an 1100 Series HPLC Value system (Agilent, Santa Clara, CA)equipped with a Gemini-NX(00F-4453-E0,Phenomenex,Torrance,CA)column(4.6 mm×150 mm,3 μm,100 A?).The collected 45 fractions were concentrated via vacuum centrifugation and reconstituted in 40 μl of 0.1% v/v trifluoroacetic acid. Then, the TMT-labeled samples were analyzed using an Easy-nLC nanoflow HPLC system (ThermoFisher Scietific), which was connected to an Orbitrap Fusion mass spectrometer (ThermoFisher Scietific).
The raw data were analyzed using Proteome Discoverer 2.1 software (ThermoFisher Scietific) with mascot 2.3 against the protein database translated from the Saccharina japonica transcriptome(GSA:PRJCA000815). The search parameters were used as described by Chen L et al. [62]. The final ratios obtained from the relative protein quantifications were normalized based on the IS and median average protein quantification ratio followed by Student’s t-test for P value.
Data availability
The raw sequence data are deposited in the Genome Sequence Archive [63] at the National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation (GSA: PRJCA000815),and are publicly accessible at http://bigd.big.ac.cn/gsa. The raw protein data and metadata are deposited in the ProteomeXchange Consortium via the iProX partner repository(ProteomeXchange:PXD009642),and are publicly accessible at http://proteomecentral.proteomexchange.org.The cDNA sequences of SjaM1PDH and SjaM1Pase genes are deposited in GenBank: SjaM1PDH1 (GenBank:MF706368), SjaM1PDH2 (GenBank: MF706369), SjaM1-Pase1 (GenBank: MF440344), and SjaM1Pase1 (GenBank:MF465902).
CRediT author statement
Shan Chi:Methodology,Formal analysis,Visualization,Writing-original draft,Writing-review&editing.Guoliang Wang:Methodology, Formal analysis, Data curation, Visualization,Software, Writing - original draft, Writing - review & editing.Tao Liu: Conceptualization, Resources, Supervision, Funding acquisition. Xumin Wang: Conceptualization, Resources,Supervision. Cui Liu:Investigation. Yuemei Jin:Investigation.Hongxin Yin: Investigation. Xin Xu: Investigation. Jun Yu:Conceptualization, Resources, Writing - review & editing,Supervision. All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 41376143), the Leading Talents Program in Taishan Industry of Shandong Province,the Leading Talents Program in Entrepreneurship and Innovation of Qingdao, the China-ASEAN Maritime Cooperation Fund ‘‘China-ASEAN Center for Joint Research and Promotion of Marine Aquaculture Technology”, the China Agriculture Research System (Grant No. CARS-50), and the Seed Industry Innovation and Industrialization Project of Fujian Province, China.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2018.12.012.
ORCID
0000-0002-5625-6457 (Shan Chi)
0000-0002-5367-2244 (Guoliang Wang)
0000-0002-7252-4300 (Tao Liu)
0000-0003-0302-8909 (Xumin Wang)
0000-0002-1935-7590 (Cui Liu)
0000-0002-2274-9237 (Yuemei Jin)
0000-0002-3161-7498 (Hongxin Yin)
0000-0002-9339-965X (Xin Xu)
0000-0002-2702-055X (Jun Yu)
 Genomics,Proteomics & Bioinformatics2020年4期
Genomics,Proteomics & Bioinformatics2020年4期
- Genomics,Proteomics & Bioinformatics的其它文章
- Global Analysis of Gene Expression Profiles Provides Novel Insights into the Development and Evolution of the Large Crustacean Eriocheir sinensis
- mrMLM v4.0.2: An R Platform for Multi-locus Genome-wide Association Studies
- Denoising Autoencoder, A Deep Learning Algorithm, Aids the Identification of A Novel Molecular Signature of Lung Adenocarcinoma
- G9a/GLP-sensitivity of H3K9me2 Demarcates Two Types of Genomic Compartments
- The Wolfiporia cocos Genome and Transcriptome Shed Light on the Formation of Its Edible and Medicinal Sclerotium
- Tissue-specific Gene Expression Changes Are Associated with Aging in Mice