
Restrictive Cardiomyopathy Resulting from a Troponin I Type 3 Mutation in a Chinese Family
2016-09-06 02:21:14YanpingRuanChaoxiaLuXiaoyiZhaoRuijuanLiangHuiLianMichaelRoutledgeWeiWuXueZhangandZhongjieFan
Chinese Medical Sciences Journal 2016年1期
Yan-ping Ruan, Chao-xia Lu, Xiao-yi Zhao, Rui-juan Liang, Hui Lian, Michael Routledge, Wei Wu, Xue Zhang*, and Zhong-jie Fan*
1Department of Cardiology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100730, China2McKusick-Zhang Center for Genetic Medicine, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100005, China3Leeds Institute of Cardiovascular & Metabolic Medicine, University of Leeds, Leeds LS2 9JZ, UK
?
Restrictive Cardiomyopathy Resulting from a Troponin I Type 3 Mutation in a Chinese Family
Yan-ping Ruan1, Chao-xia Lu2, Xiao-yi Zhao1, Rui-juan Liang1, Hui Lian1, Michael Routledge3, Wei Wu1, Xue Zhang2*, and Zhong-jie Fan1*
1Department of Cardiology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100730, China2McKusick-Zhang Center for Genetic Medicine, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100005, China3Leeds Institute of Cardiovascular & Metabolic Medicine, University of Leeds, Leeds LS2 9JZ, UK
restrictive cardiomyopathy; autosomal dominant; troponin I
Objective To identify the pathogenic variant responsible for restrictive cardiomyopathy (RCM) in a Chinese family.
Methods Next generation sequencing was used for detecting the mutation and results verified by sequencing. We used restriction enzyme digestion to test the mutation in the family members and 200 unrelated normal subjects without any cardiac inherited diseases when the mutation was identified.
Results Five individuals died from cardiac diseases, two of whom suffered from sudden cardiac death. Two individuals have suffered from chronic cardiac disorders. Mutation analysis revealed a novel missense mutation in exon 7 of troponin I type 3 (TNNI3), resulting in substitution of serine (S) with proline (P) at amino acid position 150, which cosegregated with the disease in the family, which is predicted to be probably damaging using PolyPhen-2. The mutation was not detected in the 200 unrelated subjects we tested.
Conclusion Using next generation sequencing, which has very recently been shown to be successful in identifying novel causative mutations of rare Mendelian disorders, we found a novel mutation of TNNI3 in a Chinese family with RCM.
Chin Med Sci J 2016; 31(1):1-7
R ESTRICTIVE cardiomyopathy (RCM) is a rare heart muscle disease, characterized by impaired ventricular filling with a normal or decreased diastolic volume and normal or near normal systolic function. Clinically, RCM patients present with symptoms of dyspnea, a major complaint, and sometimes with chest pain, palpitation and pulmonary edema that may be so severe as to require transplantation.1Furthermore, sudden cardiac death (SCD) is prevalent in pediatric RCM with a two-year mean survival rate for pediatric cases reaching about 50% after diagnosis.2-4Rivenes et al5and Palka et al6have confirmed the association between the ischemia and SCD in RCM. No effective treatment is available for RCM.7
During the last decade, more genes, including 4 sarcomere genes, such as troponin I type 3 (TNNI3), troponin T type 2 (TNNT2), myosin heavy chain 7 (MYH7), and actin, alpha cardiac muscle 1 (ACTC1), desmin, αB crystalline, and titin8,9have been reported to be causative for RCM. With the conventional mutation screening technologies, analysis of multiple genes is time-consuming and expensive. Growing genetic and phenotypic overlap among different cardiomyopathies adds further complexity. Next generation sequencing (NGS) technologies have removed these barriers and enabled testing of a large number of genes simultaneously.
In this paper, we use polymerase chain reaction (PCR)-based multiplex amplification followed by NGS and sequence target regions in Ion Torrent semiconductor sequencing platform, in order to determine the probable variant of a RCM family and provide a reference for clear etiology and genetic counseling.
PATIENTS AND METHODS
Participants
The proband was referred for atrial fibrillation and thrombus in left atrial appendage to a cardiologist in Peking Union Medical College Hospital. A detailed family history was obtained and a family tree was constructed. Medical records were acquired from the proband and his relatives. Their family comprised the proband and 15 relatives. Patients with RCM and their relatives were invited to participate in the study. Patients and individuals with symptoms underwent clinical examination, 12-lead electrocardiogram (ECG), and echocardiography. The patients with RCM were diagnosed by experienced cardiologists in accordance with the following echocardiographic criteria: (1) restrictive left ventricular filling pattern in the absence of obvious known cause for it, and (2) exclusion of constrictive pericarditis. The study was approved by the local research ethics committee and informed written consent was obtained from all participants aged ≥ 16 years old or from the parents for those aged <16 years old. The study was conducted in accordance with the Helsinki Declaration.
DNA extraction
Genomic DNA was extracted from peripheral blood samples using a kit from Tiangen [DP319, Tiangen Biochemical Technology (Beijing) Co., Ltd, China] from all participants. The DNA concentration and purity were analyzed by Nanodrop 1000 (Nanodrop Technologies Company, USA). The A260/A280ratio for all DNA samples was between 1.8 and 2.0. DNA quantity was detected using Qubit 2.0 fluorometer (Invitrogen, USA). DNA samples were diluted into 10 ng/μl for storage.
NGS and data analysis
The library was constructed using Ion AmpliSeq? Inherited Disease Panel kit (coverage uniformity >85%) and Ion AmpliSeq? Library kit 2.0 from Life Technologies (USA). The kit contained more than 10 000 pairs of primers in just three pools. The panel provides highly multiplexed target selection of all exons of over 300 genes (https://www. ampliseq.com) that are mutated in over 700 unique inherited diseases, according to NCBI ClinVar database (May 2012). There were 37 genes associated with inherited cardiomyopathy. The primers of overlapping amplicons covering the coding sequence region and flanking sequences (padding +25 base pairs) of each targeted gene were automatically generated by Ion AmpliSeqTMdesigner software. PCR reactions were performed according to the manufacturer’s instructions. DNA fragments from the samples were ligated with bar-coded sequencing adaptors with personal genome sequencing instrument (personal genome machine, PGM) by Ion Xpress? Barcode Adapters (Life Technologies). All DNA fragments were purified by AMPure? XP Reagent (Beckman Coulter Inc., USA). DNA concentration and fragment size for PCR products combined with the adapter were analyzed using Agilent BioAnalyzer DNA High-Sensitivity LabChip (Agilent Technologies Inc., USA). The average fragment sizes were about 300 bp in the prepared libraries. Emulsion PCR was performed by Ion OneTouch? 200 Template kit (Life Technologies) in Ion OneTouch (Life Technologies) according to the manufacturer’s instructions. Ion Sphere Particles (ISPs) with templates concentrated in the Ion OneTouch ES (Life Technologies) were completed. Concentrated ISPs were sequenced using Ion PGM? 200 Sequencing kit (Life Technologies; Thermo Fisher Scientific) on a 318 chip inIon Torrent PGM (Life Technologies; Thermo Fisher Scientific). Data from the PGM runs were processed using Ion Torrent Suite 4.0 software (Life Technologies; Thermo Fisher Scientific) to generate sequence reads. After sequence alignment and extraction of single nucleotide polymorphisms (SNPs) and insertion-deletion (InDel), all variants were filtered against dbSNP137. DNA sequences were visualized with an integrated genomics viewer. The most likely disease-causing variants were analyzed by Sanger sequencing (Shanghai, China).
Mutations are named on the basis of Human Genome Variation Society (http://www.hgvs.org/mutnomen). In accordance with the translation initiation codon ATG, the location of A in cDNA is defined as +1. TNNI3 sequence was referred to NM_000138.
Mutation validation
According to the results of NGS, we tested the most likely mutation in two patients and four healthy relatives using Sanger sequencing. The exon 7 of TNNI3 was amplified using intronic primers (3’-TCGTACTGGGAGACGTTTTG-5’, 5’-CCCGTTATCTGGCATAGTGG-3’) designed according to the genomic sequence of the gene using Primer 5. We used premix LA Taq (loading dye mix) (D336, TaKaRa, Japan) to perform polymerase chain reaction (PCR). The reaction system and condition conformed to manufacturer’s instructions.
When a mutation was identified, we found that a novel restriction enzyme site appeared, that is GGATCC, which was the recognition site of BamH I (TaKaRa). The PCR products were digested by the BamH I digest for 1 hour according to manufacturer’s instruction, followed by size fractionation using 3% agarose in the whole family. Then we tested 200 healthy and unrelated persons as controls using the same method.
In addition, we obtained the ID of protein encoded by TNNI3 from UniProt Database (http://www.uniprot.org/). Subsequently, we performed the prediction of functional effects of the nonsynonymous mutation in TNNI3 detected in the family using Polymorphism Phenotyping V2 (Poly-Phen-2, http://genetics.bwh.harvard.edu/pph2/) according to the instruction of PolyPhen-2.
RESULTS
Clinical investigations
There were 4 generations in the family, comprised 28 family members, with 14 members recruited in the studies (Fig. 1). The proband of this family, individual Ⅱ-7, had palpitations, exertional dyspnea with retrosternal pain 5 years ago, lasting for several minutes once, and relieved after rest or sublingual nitroglycerin. Last year, his exercise tolerance significantly decreased. His ECG showed atrial fibrillation, with ST-T change in widespread precordial and limb leads (Fig. 2A). Echocardiography revealed the myocardial involvement disorder, biatrial enlargement, mild mitral valve regurgitation, mild and modest tricuspid valve regurgitation, reduced systolic and diastolic function of left ventricle (left ventricular ejection fraction, LVEF 30%) and pericardial effusion (Fig. 2B). Coronary angiography did not reveal significant stenosis in coronary arteries.
The family history showed two individuals (Ⅲ-4, Ⅲ-11) who died suddenly at the age of 17 years old. IndividualⅠ-1 and Ⅱ-9 died of cardiac disorders at 39 years old, and Ⅱ-9 died from cardiac arrhythmia and heart failure at 59 years old. Individual Ⅲ-2 had shortness of breath and palpitation for 3 years. She had a history of syncope at the age of 17 years old. To date, the symptom has not reoccurred. Her echocardiography revealed biatrial and left ventricular enlargement, reduced systolic function (LVEF 30%) and restrictive diastolic function of left ventricular and pericardial effusion, with abnormal ST-T changes in ECG and no significant stenosis in coronary angiography.
Individual Ⅲ-5 felt shortness of breath and palpitation for 2 years during climbing up the stairs. Her ECG showed abnormal ST-T wave changes, but the echocardiography did not reach the diagnostic criteria of RCM. Individual Ⅲ-10 had no symptoms with aortic root broadening and mild aortic insufficiency, with ejection fraction 65% in echocardiography. Individual Ⅳ-1, Ⅳ-2, and Ⅳ-3 had no symptom and could take part in the physical activities as normal as healthy children. The clinical features of studied patients are shown in Table 1.
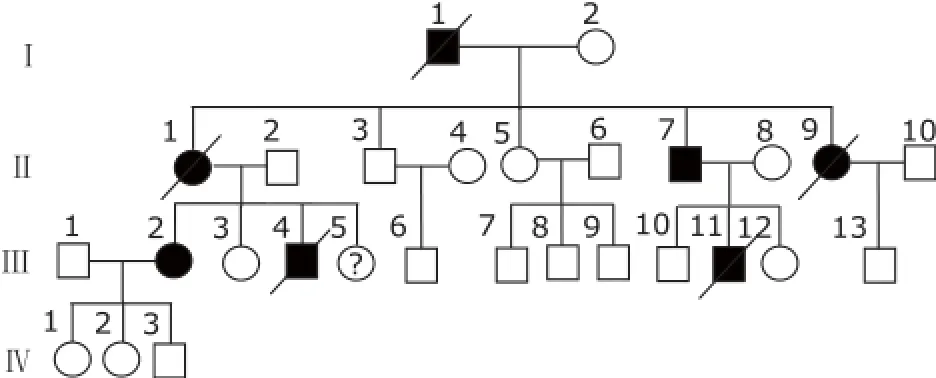
Figure 1. Pedigree drawings of the restrictive cardiomyopathy (RCM) family affected by cardiac troponin I mutation. Squares indicate male family members; circles, female family members; symbol with slash, deceased individual; open symbols, unaffected individuals; filled symbols, individuals affected by RCM; question mark, abnormal electrocardiogram, but not reaching the diagnostic criteria of RCM.
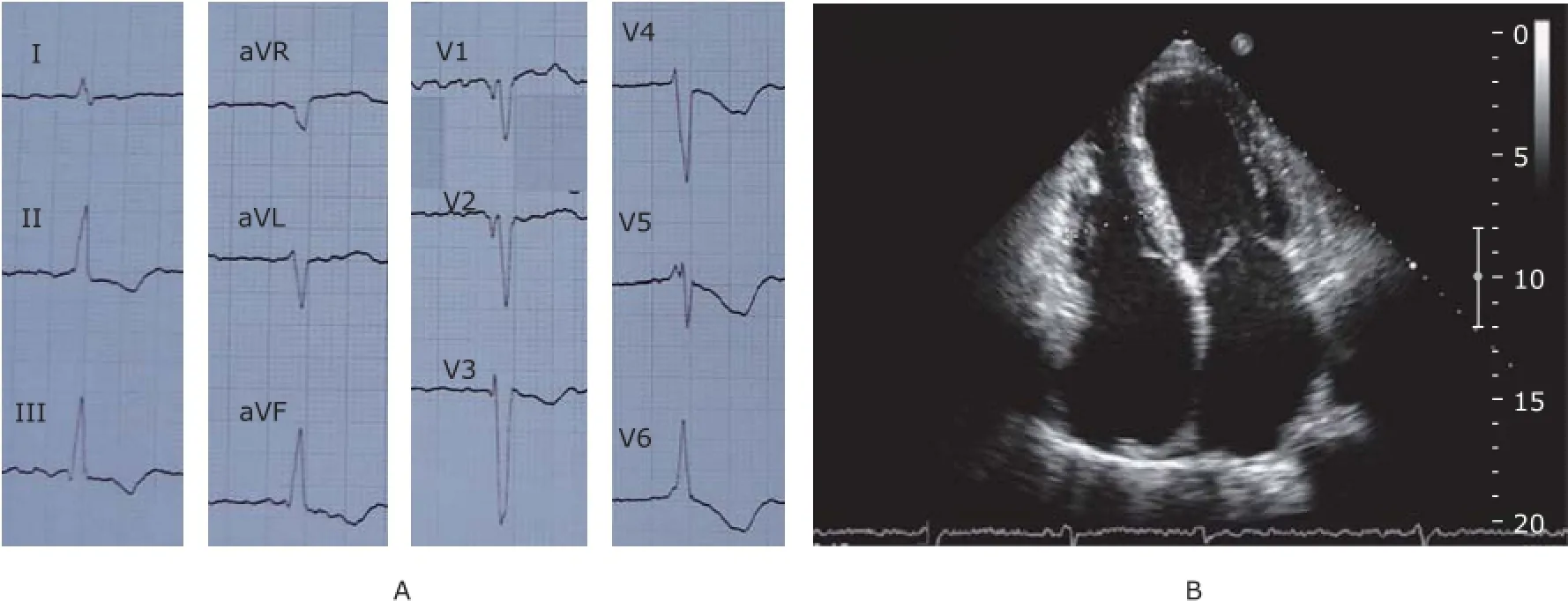
Figure 2. A. 12-lead electrocardiogram shows atrial fibrillation, ST segment depression, and T wave inversion in widespread precordial and limb leads. B. Echocardiography of individual Ⅱ-7 shows significant biatrial enlargement from apical four-chamber view.
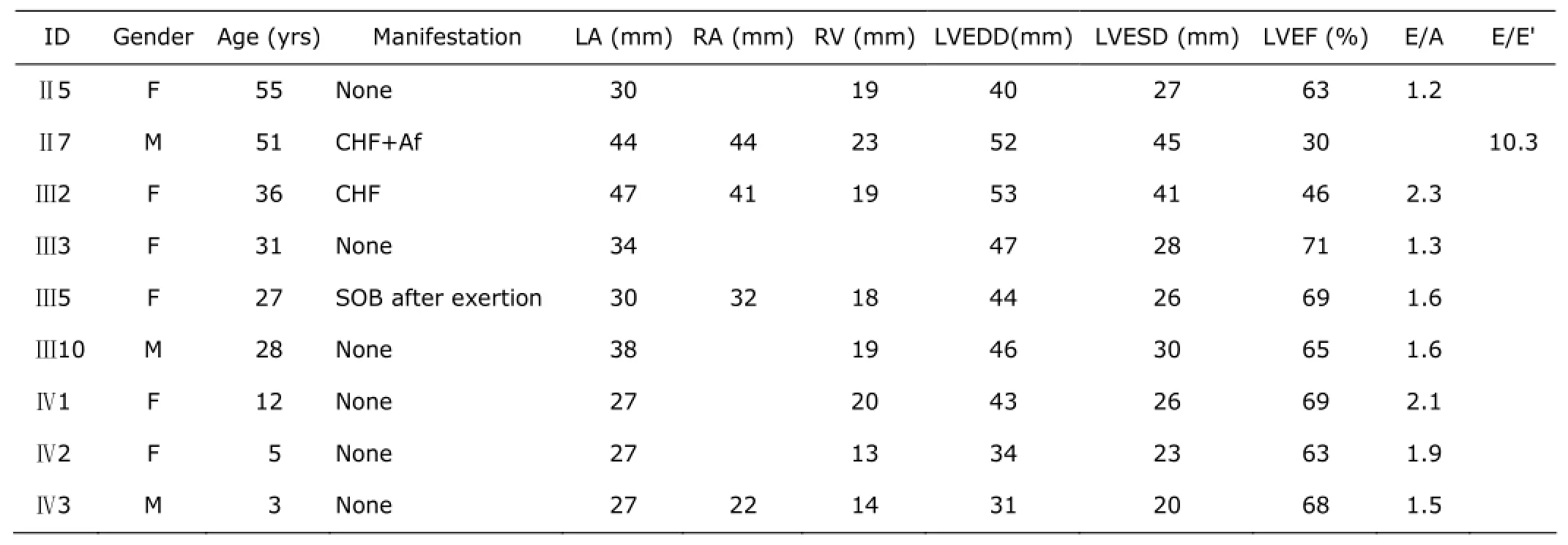
Table 1. Measurements by echocardiogram in studied objects
Genetic investigations
NGS was used to identify the gene responsible for cardiomyopathy in the proband and other family members. The results from NGS indicated that a nonsynonymous mutation c.448A>G was identified in exon 7 of TNNI3 in proband, resulting in substitution of serine (S) with proline (P) at amino acid position 150 (Fig. 3). After verifying the mutation in the patient by Sanger sequencing, we performed direct sequencing for individual Ⅱ-3, Ⅲ-2, Ⅲ-3,Ⅲ-5, Ⅲ-12, and Ⅲ-13. The results showed that Ⅲ-2 andⅢ-5 had the same mutation in exon 7 of TNNI3, while other normal individuals we tested lacked this mutation (Fig. 4). Therefore, the cosegregation between the TNNI3 marker and the disease phenotype was established in this family.
When mutation analysis of TNNI3 by direct sequencing identified a c.448A→G nucleotide substitution of exon 7, AGATCC in wild-type TNNI3 transformed into GGATCC, which was the recognition site of BamHⅠ. PCR products (447 bp) from all family members participating in our investigation underwent restriction enzyme digest for 1 hour. If the sequences contained the mutation, PCR products would be digested into 2 fragments (248 bp/199 bp). 3% agarose gel was used for size fractionation with 2000 bp DNA ladder as a marker. The PCR products from patients with RCM (Ⅱ7 and Ⅲ2) and a symptomatic family member (Ⅲ5) had been digested into 2 fragments, validating them as mutation carriers. We also observedindividual Ⅳ3, a three-year old boy, was a mutation carrier (Fig. 4). Because of autosomal dominant inheritance in the family, there were 3 bands in the gel image in these patients, while there was only 1 fragment sized 447 bp in other healthy relatives.
UniProt Database (http://www.uniprot.org/) showed the accession number of protein encoded by TNNI3 was P19429. PolyPhen-2 analysis revealed that Ser150Pro amino acid substitution is predicted to be probably damaging with a score of 0.991 (sensitivity: 0.50, specificity: 0.95). (Fig. 5).
The results of restriction enzyme digest method indicated that PCR products from 200 unrelated healthy individuals (age range 18-50 years old, 67% females) without any cardiac inherited diseases could not be digested into 2 fragments. So we can confirm that the mutation was not observed in the TNNI3 gene of any of the 200 healthy persons.
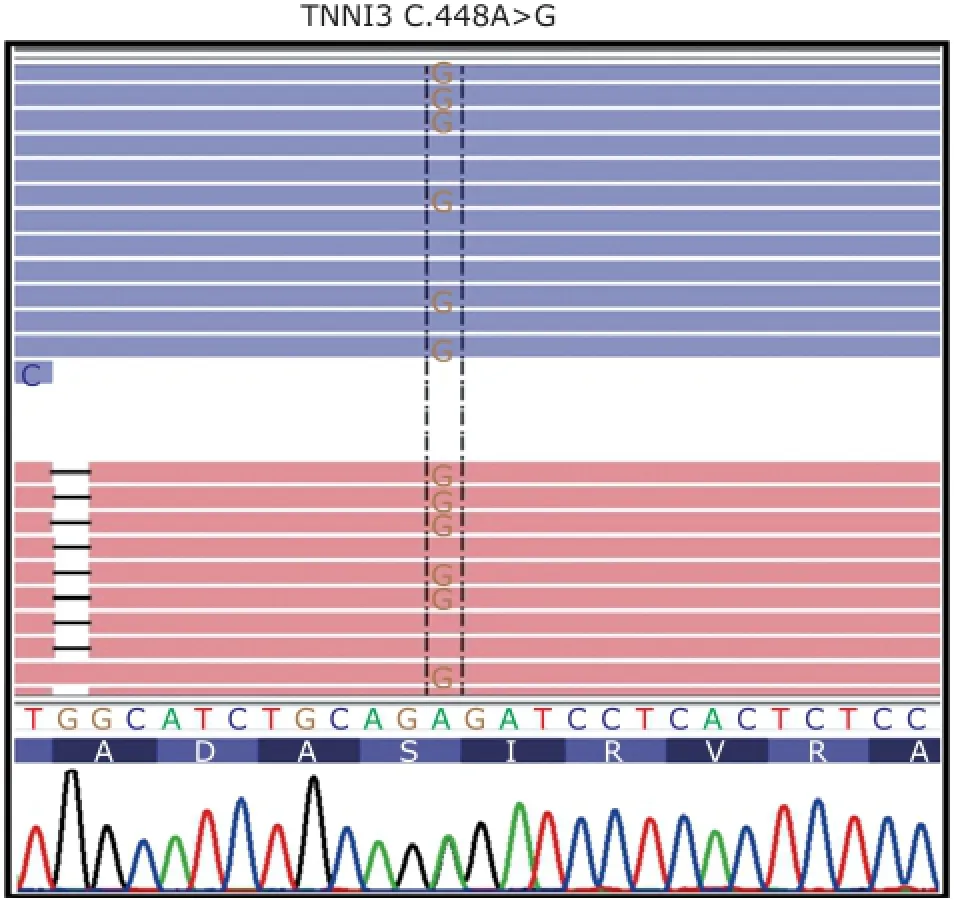
Figure 3. Partial sequence of exon 7 of troponin I type 3 (TNNI3). Mutation spot is labeled in the dotted box, that is A>G.
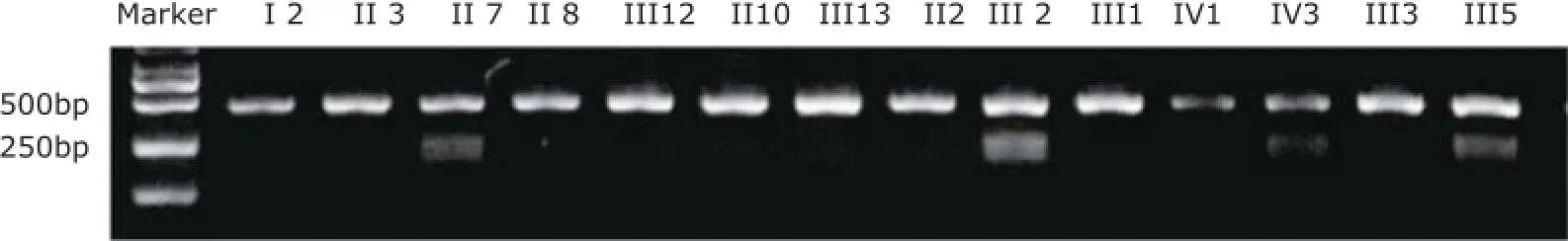
Figure 4. Electrophoretic bands of PCR production through restriction enzyme digest by gel imaging and analysis system in this family.
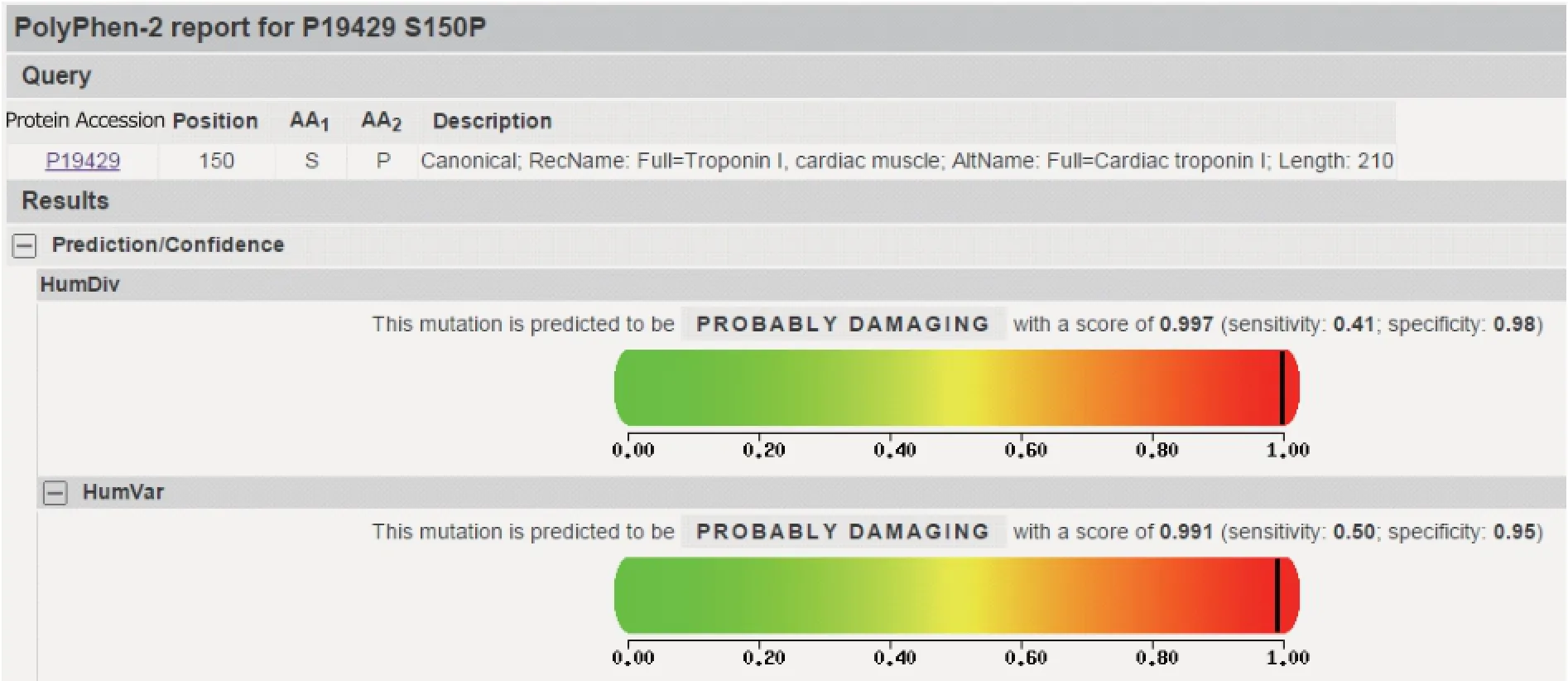
Figure 5. Prediction of functional effects of a nonsynonymous mutation c.448A>G in TNNI3 using Polymorphism Phenotyping V2.
DISCUSSION
RCM is an uncommon heart muscle disorder, characterized by impaired ventricular filling and reduced diastolic volume with or without systolic dysfunction, which may be associated with systemic disease but is most often idiopathic. Due to the poor prognosis and the high prevalence of SCD, especially in children, prenatal gene diagnosis is becoming more and more important. Therefore, many researchers focus on detecting potential pathogenic mutations associated with RCM. Over the past decade, troponin I, desmin, troponin T, cardiac beta myosin heavy chain, and ACTC1 have been established as heritable causes of RCM. It also has been shown that sarcomeric gene mutations may be associated with both hypertrophic cardiomyopathy (HCM)and dilated cardiomyopathy,10with different location within specific functional domains resulting in different phenotypes.11TNNI3 mutations were identified in a significant portion of such patients. In 2003, Mogensen et al12recognized a large family in which individuals were affected by either RCM or HCM and an additional 9 unrelated RCM patients using linkage analysis, which has determined several mutations in TNNI3, resulting in Asp190His, Arg192His, Lys178Glu, Arg145Trp, Leu144Gln amino acid substitution. These mutations has been detected in other studies.13,14In 2015, Mouton et al15have identified a novel disease-causing Leu144His mutation and a de novo Arg170Gln mutation associated with RCM and focal ventricular hypertrophy, often below the typical diagnostic threshold for HCM. In addition, Kaski et al16have performed the genetic evaluation in 12 patients with idiopathic RCM (mean age 5.1 years), which has confirmed that Lys178Glu amino acid substitution in troponin encoded by TNNI3 gene associated with RCM, and more importantly, firstly identified a novel deletion of 2 nucleotides (g.4789_ 4790delAA) in exon 7 of TNNI3, resulting in a frame shift and the introduction of a premature termination codon at amino acid position 209 (Glu177fsX209). Meanwhile, the study also showed that a nucleotide substitution in exon 10 in TNNT2 (g.9718G→A) and in exon 5 of ACTC (g.4642G→C).16What’s more, Peled et al9has identified that a de novo mutation within exon 266 of the titin gene, resulting in Tyr7621Cys amino acid substitution, which affects a highly conserved region in the protein within afibronectin-3 domain in a family affected by RCM. Most recently, Wu et al17performed gene analysis for a multigenerational family with 3 live patients and 1 unrelated patient with clinical diagnoses of RCM using NGS, which identified a nonsense mutation in myosin-binding protein C (MYBPC3) gene (c.1387C>T, p.Gln463X) and a missense MYBPC3 mutation (c.1000G>A, p.Glu334Lys). In this study, we recognized a novel mutation in TNNI3 causing RCM in a family in China, resulting in substitution of serine (S) with proline (P) at amino acid position 150, which has never been reported. What’s more, the mutation was predicted to be probably damaging according to the PolyPhen-2 report.
It is known that cardiac contractility is controlled by the thin filament regulatory proteins that form the sarcomere as a unit, including cardiac troponin, tropomyosin, myosin, and actin. Cardiac troponin complex, important in the regulation of cardiac muscle contraction, consists of 3 subunits: cardiac troponin C, as Ca2+sensor; troponin I, the inhibitory subunit, primarily functioning to prevent actin and myosin from interacting in the absence of Ca2+; and troponin T binding to tropomyosin and responsible for transmitting the Ca2+binding signal from troponin to tropomyosin. Muscle contraction is initiated by binding of calcium, released from the sarcoplasmic reticulum, to troponin C. This results in a conformational change of the troponin complex attenuating the inhibitory effects of troponin I on actomyosin crossbridge formation. TNNI3 contains 8 exons, which encode a basic globular protein containing approximately 210 amino acids (24 kDa), including 3 domains: residue 61-112 is a troponin T binding domain; residue 113-164 is a troponin C binding domain and residue 130-148, 173-181 is an actin binding domain.12We identified the substitution of serine (S) with proline (P) at amino acid position 150 in our patients, which is in the troponin C binding domain. When Ca2+decreases, the configuration of cardiac troponin C and troponin I returns to the status before constriction, and cardiac troponin I combines with actin again. But the process cannot be completed once mutation in this location has occurred. Therefore, the diastolic filling was impaired in patients affected with RCM.
Studies in vitro have shown that increase in Ca2+sensitivity may be the pathogenic mechanism of RCM in patients with TNNI3 mutations.18,19In 2009, Wen and colleagues20performed functional effects of a RCM linked cardiac troponin I mutation (Arg145Trp). The results revealed that a ~13%-16% increase in maximal Ca2+-activated force and ATPase activity compared with troponin I wild-type transgenic mice, and a large increase in the Ca2+sensitivity. This resulted in impaired ventricular filling during diastole and increase in force during systole.
We used NGS to identify the gene and specific mutation of the family affected with RCM. The method has revolutionized the genomic and transcriptomic approaches to biology by reducing the sequencing cost and significantly increasing the throughput. NGS has very recently been shown to be successful in identifying novel causative mutations of rare or common Mendelian disorders, although the method in the field of cardiovascular diseases has not yet been applied widely.21,22Meanwhile, the development of NGS devices and program are now providing the solid foundation for researchers to identify the single-gene or multi-gene inherited diseases. The probably damaging mutation in TNNI3 associated with RCM detected in this family may guide the young mutation carriers to avoid the vigorous activities, periodically monitor the change of echocardiography and ECG to initiate early treatment, and more importantly, help them perform gene diagnosis early in pregnancy,
Amino acid abbreviations used
Arg: arginine; Asp: aspartic acid; His: histidine; Lys: lysine;Glu: glutamic acid; Trp: tryptophan; Leu: leucine; Gln: glutamine; Tyr: tyrosine; Cys: cysteine.
ACKNOWLEDGEMENTS
We appreciate the contribution of staffs in the genetic laboratory in Peking Union Medical College, who have given great support for the technical problems during the study. All the authors thank the subjects for participating in the study.
REFERENCES
1. Willott RH, Gomes AV, Chang AN, et al. Mutations in troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function. J Mol Cell Cardiol 2010; 48:882-92.
2. Kimberling MT, Balzer DT, Hirsch R, et al. Cardiac transplantation for pediatric restrictive cardiomyopathy: presentation, evaluation, and short-term outcome. J Heart Lung Transplant 2002; 21:455-9.
3. Peddy SB, Vricella LA, Crosson JE, et al. Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene. Pediatrics 2006; 117:1830-3.
4. Greenway SC, Wilson GJ, Wilson J, et al. Sudden death in an infant with angina, restrictive cardiomyopathy, and coronary artery bridging: an unusual phenotype for a beta-myosin heavy chain (MYH7) sarcomeric protein mutation. Circ Heart Fail 2012; 5:e92-3.
5. Rivenes SM, Kearney DL, Smith EO, et al. Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation 2002; 102:876-82.
6. Palka P, Lange A, Ward C. A fatal case of idiopathic restrictive cardiomyopathy. Cardiol Young 2003; 13:469-71.
7. Jean-Charles PY, Li YJ, Nan CL, et al. Insights into restrictive cardiomyopathy from clinical and animal studies. J Geriatr Cardiol 2011; 8:168-83.
8. Parvatiyar MS, Pinto JR, Dweck D, et al. Cardiac troponin mutations and restrictive cardiomyopathy. J Biomed Biotechnol 2010; 2010:350706.
9. Peled Y, Gramlich M, Yoskovitz G, et al. Titin mutation in familial restrictive cardiomyopathy. Int J Cardiol 2014; 171:24-30.
10. Kamisago M, Sharma SD, DePalma SR, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med 2003; 343:1688-96.
11. Chen J, Chien KR. Complexity in simplicity: monogenic disorders and complex cardiomyopathies. J Clin Invest 1999; 103:1483-5.
12. Mogensen J, Kubo T, Dugue M, et al. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest 2003; 111:209-16.
13. Chen Y, Yang S, Li J, et al. Pediatric restrictive cardiomyopathy due to a heterozygous mutation of the TNNI3 gene. J Biomed Res 2014; 28:59-63.
14. Yang SW , Chen Y , Li J , et al. Clinical characteristics and genetic analysis of three pediatric patients with idiopathic restrictive cardiomyopathy. Zhonghua Xin Xue Guan Bing Za Zhi 2013; 41:304-9.
15. Mouton JM, Pellizzon AS, Goosen A, et al. Diagnostic disparity and identification of two TNNI3 gene mutations, one novel and one arising de novo, in South African patients with restrictive cardiomyopathy and focal ventricular hypertrophy. Cardiovasc J Afr 2015; 26:63-9.
16. Kaski JP, Syrris P, Burch M, et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 2008; 94:1478-84.
17. Wu W, Lu CX, Wang YN, et al. Novel phenotype-genotype correlations of restrictive cardiomyopathy with myosinbinding protein C (MYBPC3) gene mutations tested by next-generation sequencing. J Am Heart Assoc 2015; 4. pii: e001879.
18. Gomes AV, Liang J, Potter JD. Mutations in human cardiac troponin I that are associated with restrictive cardiomyopathy affect basal ATPase activity and the calcium sensitivity of force development. J Biol Chem 2005; 280:30909-15.
19. Yumoto F, Lu QW, Morimoto S, et al. Drastic Ca2+sensitization of myofilament associated with a small structural change in troponin I in inherited restrictive cardiomyopathy. Biochem Biophys Res Commun 2005; 338:1519-26.
20. Wen Y, Xu Y, Wang Y, et al. Functional effects of a restrictive-cardiomyopathy-linked cardiac troponin I mutation (R145W) in transgenic mice. J Mol Biol 2009; 392: 1158-67.
21. Davey JW, Hohenlohe PA, Etter PD, et al. Genome-wide genetic marker discovery and genotyping using nextgeneration sequencing. Nat Rev Genet 2011; 12:499-510.
22. Faita F, Vecoli C, Foffa I, et al. Next generation sequencing in cardiovascular diseases. World J Cardiol 2012; 4: 288-95.
for publication August 18, 2015.
*Corresponding author Zhong-jie Fan, Tel: 86-10-69155086, E-mail: fan@pumch.cn, Xue Zhang, E-mail: xuezhang@pumc.edu.cn
 Chinese Medical Sciences Journal2016年1期
Chinese Medical Sciences Journal2016年1期
- Chinese Medical Sciences Journal的其它文章
- Pathology Verified Concomitant Papillary Thyroid Carcinoma in the Sonographically Suspected Thyroid Lymphoma: A Case Report△
- Life-threatening Spontaneous Retroperitoneal Haemorrhage: Role of Multidetector CT-angiography for the Emergency Management
- Percutaneous Removal of Benign Breast Lesions with an Ultrasound-guided Vacuum-assisted System: Influence Factors in the Hematoma Formation△
- Respiratory and Cardiac Characteristics of ICU Patients Aged 90 Years and Older: A Report of 12 Cases
- Establish Albumin-creatinine Ratio Reference Value of Adults in the Rural Area of Hebei Province△
- Positive Rate of Different Hepatitis B Virus Serological Markers in Peking Union Medical College Hospital, a General Tertiary Hospital in Beijing△